




El desarrollo de modelos animales de una enfermedad ha sido siempre bien acogido por la comunidad científica porque permite realizar una aproximación a la investigación de determinados aspectos de la misma.
Los modelos animales de la EPOC no pueden llegar a reproducir la heterogeneidad de esta enfermedad y generalmente solo llegan a representar los estadios más leves de la misma. Además, la obstrucción al flujo aéreo, variable que determina el diagnóstico en un paciente, no siempre se tiene en cuenta en los modelos. Por este motivo, los modelos se han centrado en el desarrollo de enfisema, fácilmente detectable por morfometría pulmonar, sin prestar atención a otros componentes de la enfermedad, como la lesión de las vías aéreas o las alteraciones vasculares asociadas.
La exposición continua y prolongada al humo de tabaco se considera el principal factor de riesgo de esta enfermedad, lo que justifica que sea el modelo de exposición al humo de tabaco el más ampliamente utilizado. Sobre esta base de modelo podemos encontrar algunas variantes relacionadas con el tiempo de exposición, la asociación de otros inductores o inhibidores, las exacerbaciones o el uso de animales transgénicos que facilitan la identificación de las vías patogénicas. Es posible, por tanto, reproducir algunas variantes o heterogeneidades de esta enfermedad y diseñar uno u otro modelo que sea capaz de responder a una u otra pregunta de investigación, dirigida bien a una identificación patogénica y/o bien a una respuesta terapéutica.
Animal models of disease have always been welcomed by the scientific community because they provide an approach to the investigation of certain aspects of the disease in question.
Animal models of COPD cannot reproduce the heterogeneity of the disease and usually only manage to represent the disease in its milder stages. Moreover, airflow obstruction, the variable that determines patient diagnosis, not always taken into account in the models. For this reason, models have focused on the development of emphysema, easily detectable by lung morphometry, and have disregarded other components of the disease, such as airway injury or associated vascular changes.
Continuous, long-term exposure to cigarette smoke is considered the main risk factor for this disease, justifying the fact that the cigarette smoke exposure model is the most widely used. Some variations on this basic model, related to exposure time, the association of other inducers or inhibitors, exacerbations or the use of transgenic animals to facilitate the identification of pathogenic pathways have been developed. Some variations or heterogeneity of this disease, then, can be reproduced and models can be designed for resolving researchers’ questions on disease identification or treatment responses.
El elevado impacto de la enfermedad pulmonar obstructiva crónica (EPOC) a nivel mundial obliga a disponer de todas las herramientas posibles para abordar todos los aspectos de la enfermedad. Los problemas del infradiagnóstico, del paciente frágil exacerbador o las incógnitas existentes acerca del desarrollo de una u otra forma clínica de la enfermedad o de la historia natural de la misma, en la que se mezclan casos de muy poca progresión con casos de progresión acelerada, pueden ser abordables bajo el desarrollo de modelos animales. Del mismo modo, todo nuevo ensayo terapéutico generalmente comienza con una aproximación realizada previamente en un modelo animal.
El tabaquismo es la principal causa de esta enfermedad, pero su capacidad para generar una respuesta inflamatoria permanente depende de la susceptibilidad del paciente. Por eso, los modelos animales de la EPOC desarrollados por exposición al humo del tabaco son los principalmente elegidos para estudiar los mecanismos patogénicos de la enfermedad y de la susceptibilidad a su desarrollo y progresión. El uso de animales transgénicos que tienen limitada o potenciada alguna vía metabólica ayuda en estos casos a comprender las vías patogénicas existentes en uno u otro caso. Igualmente, dentro del estudio de esta enfermedad, las nuevas aproximaciones a la clasificación de la EPOC que proponen tanto la estrategia GOLD1 como la GESEPOC2 dan mayor importancia a la existencia de las exacerbaciones por la trascendencia que tienen sobre la intensidad de los síntomas, la progresión de la obstrucción y la mortalidad. Por este motivo, el estudio de las exacerbaciones en los modelos de la EPOC ha cobrado mayor interés y pueden contribuir a mejorar el conocimiento acerca de los mecanismos que subyacen en la condición del paciente exacerbador.
Los resultados encontrados con el uso de modelos animales de cualquier enfermedad deben siempre considerarse bajo la limitación de tener que extrapolar una conclusión sobre lo potencialmente existente en un paciente, pero son imprescindibles como soporte de la investigación clínica si se plantean como modelos preclínicos, término cada vez más utilizado, y que engloba el concepto de traslación a la clínica y que debe prevalecer en todo diseño experimental.
Modelos de la enfermedad pulmonar obstructiva crónica por exposición al humo de tabacoLos modelos de la EPOC por exposición al humo de tabaco son los que mejor reflejan los mecanismos inflamatorios y patogénicos de la enfermedad y, por tanto, los que potencialmente mejor se prestan a los ensayos de nuevas terapias. La exposición al humo de tabaco se ha aplicado en numerosas especies animales, como perros, cobayas, conejos, ratas y ratones. De ellas, las cobayas y los ratones han demostrado ser las especies más susceptibles para el desarrollo de la enfermedad por exposiciones prolongadas3. Existen 2 procedimientos generales de administración de humo de tabaco: el denominado «nose only», donde la combustión del cigarrillo es conducida directamente a la nariz del animal, y el «whole body» (fig. 1), donde el animal es expuesto íntegramente en una cámara sometida a una concentración controlada de humo para asegurar unos niveles estables, no tóxicos, de carboxihemoglobina4,5. Conceptualmente diferentes, ambos métodos se han utilizado indistintamente y se han mostrado similares en cuanto a los hallazgos referentes a la presencia de poblaciones celulares inflamatorias, niveles de citoquinas, cambios en el remodelado pulmonar y respuestas terapéuticas6.

Aproximadamente el 90% de los pacientes que sufren la EPOC es por haber fumado un promedio de al menos 10 paquetes/año y desarrollan una enfermedad que puede presentarse bajo diferentes formas clínicas con diferentes niveles de progresión y gravedad de las mismas7. El modelo animal de la EPOC, sea de cobaya o murino, suele establecerse durante un periodo de exposición de 6meses8, aunque a partir del segundo mes ya se detectan importantes cambios inflamatorios y morfométricos (fig. 2)9. Nunca suelen alcanzar las fases equivalentes a la EPOC grave de un paciente, pero son capaces de desarrollar muchas de las características propias de esta enfermedad, como inflamación crónica con afluencia de neutrófilos y macrófagos, presencia de linfocitosT CD4 y CD8, hipersecreción de moco, cambios en la función pulmonar, enfisema y remodelado vascular y de las vías aéreas10.
 del pulmón de ratones expuestos al aire ambiental (A) y de ratones expuestos durante 6 meses al humo del tabaco (B), mostrando enfisema.")
El modelo murino de exposición al humo de tabaco es el más utilizado debido a su bajo coste y fácil manejo, al buen conocimiento de su genoma, a la disponibilidad de muchas variantes transgénicas, a la disponibilidad de una variada oferta de anticuerpos específicos para su uso en el laboratorio y a la disponibilidad de una amplia gama de cepas que ofrecen diferentes susceptibilidades en su respuesta al humo de tabaco. La susceptibilidad para desarrollar la EPOC dependiente de cepa está bien identificada en el modelo murino11–14, y si la exposición de ratones al humo de tabaco se realiza durante al menos 3 a 6meses, se puede generar el patrón morfométrico pulmonar de la EPOC con presencia de células inflamatorias típicas, mediadores de inflamación y cambios funcionales propios de la enfermedad15.
Los cobayas también presentan algunas ventajas a la hora de generar modelos de la EPOC, ya que son animales muy susceptibles para desarrollar la enfermedad tras pocos meses de exposición16. En 1990, Wright y Churg17 publicaron uno de los primeros estudios con cobayas expuestas al humo de tabaco. En este caso, tras 12meses de exposición las cobayas habían desarrollado enfisema y presentaban alteraciones de la función pulmonar de forma muy similar a lo que ocurre en fumadores con la EPOC. La principal limitación en el uso de esta especie proviene de la limitada disponibilidad de anticuerpos específicos. Las ratas, especie de roedores próxima a ratones y cobayas, son más resistentes para desarrollar alteraciones por exposición al humo de tabaco, por lo que su uso como modelo de la EPOC es menos frecuente18. La tabla 1 resume algunos hallazgos relevantes descritos en este modelo.
Ejemplos de modelos de la EPOC por exposición al humo de tabaco
| Modelo animal | Metodología | Aportación | Referencia |
|---|---|---|---|
| Ratón Serpina B1 KO | Exposición crónica | No se asocia con enfisema severo | 83 |
| Ratón NZWLac/J, AJ, SJL, C57BL/6 y AKR | Exposición crónica | El grado de susceptibilidad a desarrollar daño pulmonar varía entre distintas cepas | 12 |
| Ratón NZWLac/J y AKR | Exposición crónica | Egr-1 (marcador pro-inflamatorio) altamente incrementado en la cepa susceptible AKR, mientras que en la cepa NZWLac/J el incremento fue mínimo | 84 |
| Ratón C57BL/6 (CD8−/−) | Exposición crónica | Los linfocitos T CD8+ son imprescindibles para el desarrollo de enfisema | 85 |
| Ratón C57BL/6 | Exposición aguda | Aumento de marcadores de daño sobre el ADN por estrés oxidativo (8-ohdg y 4-HNE) | 86 |
| Ratón ICR (Nrf2−/−) | Exposición crónica | Más susceptibles a desarrollar enfisema, con incremento de la expresión de enzimas antioxidantes | 78 |
| Cobaya | Exposición crónica | Aumento del remodelado vascular y de la presión pulmonar arterial | 87 |
| Ratón C57BL/6 y «pallid» | Exposición crónica | Los niveles de α1-antitripsina determinan el perfil enfisematoso | 88 |
| Cobaya | Exposición crónica | Papel de las MMP en el desarrollo de enfisema y remodelado de las vías aéreas | 89 |
| Ratón C57BL/6 | Exposición aguda (directa e indirecta) | La intensidad de la respuesta inflamatoria depende de la composición del humo de tabaco | 90 |
| Ratón C57BL/6 (sGCα1(−/−)) | Exposición aguda y crónica | Reducción de la expresión de sGC contribuyendo a la limitación al flujo aéreo | 91 |
| Ratón | Exposición subcrónica | Efecto antiinflamatorio de los ligandos de PPAR-γ | 92 |
| Ratón | Exposición crónica | La neutralización de CXCL13 bloquea parcialmente la respuesta inflamatoria y la destrucción de la pared alveolar | 93 |
| Ratón | Exposición crónica | El bloqueo de linfocitosT puede ser eficaz como terapia para la EPOC | 94 |
| Ratón C57BL/6 y BALB/cJ | Exposición crónica | El humo de tabaco desencadena una respuesta dependiente de antígeno en la que participan linfocitos T CD4+ y CD8+ | 95 |
| Ratón C57BL/6 y 129S2/SvHsd | Exposición aguda | Implicación de los monocitos proinflamatorios en la susceptibilidad a desarrollar daño pulmonar | 5 |
Los modelos de la EPOC por exposición crónica al humo de tabaco siguen teniendo la limitación de que se muestran incapaces de reproducir algunas de las características de esta enfermedad compleja y heterogénea. Formas clínicas conocidas, como la del exacerbador, de progresión acelerada o con colonización bacteriana, por ejemplo, no han podido ser desarrolladas hasta la fecha, aunque existen algunas aproximaciones mediante la combinación de agentes que intentan dar respuesta a este planteamiento. La exposición a gases tóxicos e irritantes como el dióxido de nitrógeno, el ozono o el dióxido de azufre produce una lesión pulmonar más intensa que la del humo de tabaco19–22.
Modelos de exacerbaciones de la enfermedad pulmonar obstructiva crónicaLa presencia de exacerbaciones es una característica de la EPOC que si se presenta de manera repetida condiciona una peor evolución de los pacientes por asociarse a una mayor progresión de la enfermedad, peor calidad de vida y mayor mortalidad. La posibilidad de disponer de modelos animales de exacerbación abre la opción de estudiar mecanismos patogénicos asociados y detectar posibles marcadores biológicos asociados.
La mayoría de las exacerbaciones infecciosas de la EPOC son de origen viral (75%), seguidas de las de origen bacteriano (25%). Disponemos de datos de modelos in vivo acerca del efecto de la infección viral sobre ratones previamente expuestos al humo de tabaco, de forma tanto aguda como crónica. El pulmón responde con mayor intensidad de inflamación si la infección viral afecta a un animal previamente expuesto al humo de tabaco. Además, se acelera la progresión del enfisema y el nivel de afectación de las vías aéreas23,24.
La bacteria más comúnmente aislada en las exacerbaciones de la EPOC es el Haemophilus influenzae no tipificable (HiNT). Por ello tiene especial interés el resultado obtenido en modelos de esta infección sobre ratones sanos25 o previamente expuestos al humo de tabaco. Tras 8semanas de exposición a tabaco en ratones de la cepa C57BL/6, la infección con HiNT produce una respuesta inflamatoria más intensa y un daño pulmonar mayor que en animales previamente sanos26,27.
Los lipopolisacáridos bacterianos se han utilizado solos, en administración crónica28 o en combinación con periodos cortos de exposición al humo de tabaco29 para desarrollar modelos de enfisema, pero administrados de manera aguda a dosis mayores pueden producir una respuesta inflamatoria que se acompaña de fiebre, hipersecreción mucosa y broncoconstricción, que reproduce síntomas de una exacerbación30 detectables por tomografía computarizada31.
Modelos de la enfermedad pulmonar obstructiva crónica grave por combinación de agentes de inducciónEn los estadios más graves de la EPOC se produce una clara ruptura del «programa de mantenimiento» de la estructura del pulmón que puede llevar irremediablemente a la aparición de enfisema e hipertensión pulmonar. Existen varios modelos de patología de la EPOC frágil (muy grave), como la combinación de la exposición al humo de cigarrillo y el inhibidor de VEGF32. La exposición al humo de tabaco causa una disminución significativa de la expresión de VEGF y su receptor VEGFR2 en modelos animales de enfisema. Además, el tratamiento con el bloqueante del receptor de VEGF, SU5416, induce la apoptosis de las células alveolares, la retracción de los vasos capilares y el ensanchamiento del espacio alveolar33. Por ello, el enfisema aparece como un estado de deficiencia de VEGF que compromete la supervivencia de las células endoteliales y, por lo tanto, el programa de mantenimiento del pulmón. Otra aproximación puede obtenerse mediante la combinación de la exposición al humo de tabaco y la inducción de hipoxia. Este modelo puede conducir a la hipertensión pulmonar, condición presente solo en la EPOC grave evolucionada34.
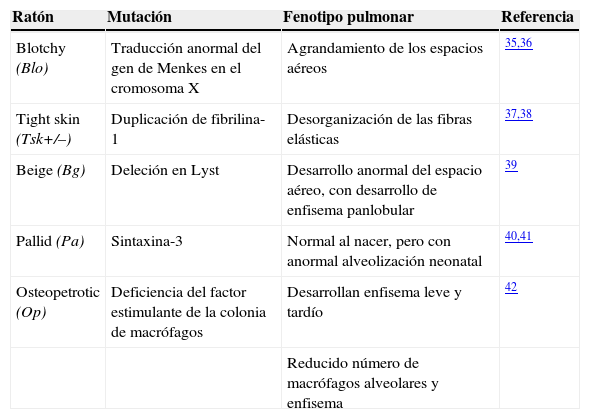
Modelos transgénicos de la enfermedad pulmonar obstructiva crónicaAntes de la llegada de la ingeniería genética dirigida ya existían algunas cepas mutantes de ratones C57BL/6 caracterizadas por desarrollar enfisema espontáneo (tabla 2). Se trata de los ratones «Blotchy», que tienen una traducción anormal del gen de Menkes en el cromosoma X35, presentando deficiencias en las proteínas del tejido conectivo pulmonar, lo que afecta a la estructura y función del pulmón con el consiguiente desarrollo de enfisema36; los «Tight Skin», que presentan una mutación en la fibrilina-1, uno de los principales componentes de las microfibrillas de la matriz extracelular del pulmón, lo que promueve el estrés oxidativo y la muerte celular, cascadas de lesiones fundamentales para el desarrollo de enfisema37,38; los «Beige», cuyos pulmones parecen normales al nacer pero que por deleción de Lyst no forman alvéolos con normalidad durante el desarrollo39; los ratones «Pallid»40, que presentan una mutación que afecta a la sintaxina-13 (una proteína de la membrana celular), que da lugar al desarrollo gradual y progresivo del enfisema41; y más recientemente los ratones «Osteopetrotic», que presentan una deficiencia del factor estimulante de la colonia de macrófagos (MCSF) y acaban desarrollando enfisema42.
Cepas de ratones mutantes naturales que desarrollan enfisema
| Ratón | Mutación | Fenotipo pulmonar | Referencia |
|---|---|---|---|
| Blotchy (Blo) | Traducción anormal del gen de Menkes en el cromosoma X | Agrandamiento de los espacios aéreos | 35,36 |
| Tight skin (Tsk+/–) | Duplicación de fibrilina-1 | Desorganización de las fibras elásticas | 37,38 |
| Beige (Bg) | Deleción en Lyst | Desarrollo anormal del espacio aéreo, con desarrollo de enfisema panlobular | 39 |
| Pallid (Pa) | Sintaxina-3 | Normal al nacer, pero con anormal alveolización neonatal | 40,41 |
| Osteopetrotic (Op) | Deficiencia del factor estimulante de la colonia de macrófagos | Desarrollan enfisema leve y tardío | 42 |
| Reducido número de macrófagos alveolares y enfisema |
Uno de los avances biotecnológicos más importantes de las últimas décadas ha sido el desarrollo de los animales transgénicos, a los que en las primeras fases del embrión se les introduce mediante inyección intranuclear un gen que no forma parte de su genoma y que secuenciará una determinada vía de interés13,15,43–45. Una de las primeras aplicaciones de la tecnología transgénica a la EPOC fue la sobreexpresión constitutiva de la colagenasa-1 (MMP-1) humana en ratones, que provoca enfisema46 por destrucción del colágeno tipoiii de la pared alveolar47. La expresión constitutiva de transgenes tiene el inconveniente de no discernir el proceso del propio desarrollo pulmonar. Para solventarlo, se desarrolló la construcción de transgénicos de expresión inducible. Así, la sobreexpresión de IL-1348, una citoquina producida por los linfocitos T-helper2 (Th2), o la sobreexpresión de IFN-γ49, producto destacado de los linfocitos T-helper1 (Th1), representan 2 ejemplos importantes de transgenes condicionales inducibles, que conducen a un enfisema en ratones adultos50, dependiente de MMP-9 y MMP-12, en el caso de los transgénicos IL-13 («Dutch»). En estos ratones la IL-13 se sobreexpresa solo cuando los animales son expuestos a la tetraciclina, permitiendo así que los investigadores activen la sobreexpresión después del desarrollo completo del pulmón. La activación del TGF-β mediada por MMP-9 parece ser la responsable de la remodelación del colágeno en este modelo. Sin embargo, en los transgénicos IFN-γ («British») el componente inflamatorio parece ser más sutil, con apoptosis prominente pero sin ninguna patología de las vías respiratorias asociada. Estos son solo 2 ejemplos que demuestran la complejidad de las redes inflamatorias y cómo hallazgos inesperados en modelos animales han conducido a la búsqueda de nuevos mediadores potenciales en la enfermedad humana. Otros trabajos indican que la inducción del TNF-α en el pulmón adulto promueve la formación de tejido linfoide y enfisema, proporcionando un modelo en el que los efectos patógenos del TNF-α en el pulmón pueden ser investigados51, o cómo la sobreexpresión de protimosina-α (ProT-α) contribuye a la patogénesis del enfisema mediante el aumento de la acetilación de histonas y expresión de MMP-2,-9 dependiente de NF-κB, sobre todo después de la exposición a tabaco52.
Un modelo alternativo al transgénico es el de «knock-out» (tabla 3), en el cual hay una inhibición en la expresión de un gen determinado, lo que permite conocer la función de las proteínas dependientes de este gen. Unas veces la inhibición génica protege contra el desarrollo de enfisema, como es el caso de la inhibición de la expresión de MMP-12, que protege del desarrollo de enfisema al tener deteriorado el sistema de reclutamiento de macrófagos al pulmón53. Otro ejemplo sería el de la ausencia de la elastasa neutrofílica (NE), que parece proteger también del desarrollo de enfisema inducido por el consumo de tabaco54. En ambos casos se demuestra el papel directo de estas proteínas en el enfisema y se pone de relieve la interdependencia de las proteinasas y las células inflamatorias que median la destrucción pulmonar en respuesta al humo del cigarrillo. Otras veces la deleción interfiere con la alveogénesis. Así, ratones que no expresan el factor de crecimiento derivado de plaquetas-A (PDGF-A) pierden miofibroblastos, y por lo tanto disminuyen los depósitos de elastina produciendo enfisema55. Los dobles knock-out para los receptores del factor de crecimiento de fibroblastos-3 y -4 (FGFR-3 y -4) muestran una formación alveolar y tabicación anormal56. Y los ratones deficientes de elastina presentan pocos y dilatados sacos aéreos distales y el desarrollo de las vías aéreas detenido57. Y en otras ocasiones la deleción de determinados genes produce agrandamiento de los espacios alveolares, como aquellos ratones que no expresan la integrina alfa(v)beta6 (integrina αVβ6), que no son capaces de activar el TGF-β en los espacios aéreos y desarrollan enfisema dependiente de MMP-1258. O los ratones knock-out para la proteínaD del surfactante pulmonar (SP-D), que presentan activación de macrófagos, producción de MMPs y agrandamiento de los espacios aéreos59. O la deficiencia del inhibidor de metaloproteasas (TIMP-3), que parece combinar el agrandamiento de los espacios aéreos con el desarrollo progresivo de enfisema60.
Ejemplos de modelos knock-out de ratones enfisematosos
| Gen | Fenotipo pulmonar | Referencia |
|---|---|---|
| Elastasa macrofágica (MMP-12) | Protección completa frente al desarrollo de enfisema tras exposición al humo del tabaco | 53 |
| Elastasa neutrofílica (NE) | Protección frente al desarrollo de enfisema inducido por el consumo de tabaco | 54 |
| Elastina | Pocos y dilatados sacos aéreos distales y desarrollo de las vías aéreas detenido | 57 |
| Factor de crecimiento derivado de plaquetas-A (PDGF-A) | Ausencia de tropoelastina y fallo en la tabicación alveolar | 55 |
| Receptor factor crecimiento fibroblastos (FGFR-3 y -4) | Formación alveolar y tabicación anormales | 56 |
| Integrina alfa(v)beta6 (integrina αvβ6) | Desarrollo espontáneo de enfisema dependiente de MMP-12 | 58 |
| Proteína D del surfactante pulmonar (SP-D) | Activación de macrófagos, producción de MMP y consecuente enfisema | 59 |
| Inhibidor de metaloproteasas (TIMP-3) | Agrandamiento de los espacios aéreos con el desarrollo progresivo de enfisema | 60 |
Para salvar la barrera trans-especie del modelo murino la tecnología también permite la eliminación selectiva de determinados genes murinos, seguido de la inserción («knocking in») de genes humanos bajo el control de promotores murinos. Existen los llamados ratones propensos al desarrollo de enfisema, a los que se les han eliminado los genes murinos de A1AT y han sido reemplazados por genes, ya sean normales o deficientes de A1AT humana61.
Modelos autoinmunes de la enfermedad pulmonar obstructiva crónicaLa inflamación pulmonar existente en la EPOC grave incluye un gran número de linfocitosT Th1 activados, linfocitos B y linfocitosT CD8, que persisten durante años, incluso después de cesar el hábito de fumar, lo que sugiere un proceso de autoperpetuación que es una de las características de las enfermedades autoinmunes. Esta cadena de eventos sugiere que la respuesta inmune adaptativa en la EPOC, junto con su persistencia después de dejar de fumar, podría ser debida a una respuesta a autoantígenos. Tras un período inicial en el que esta posibilidad solo se planteaba como hipótesis62–64, se han dado a conocer datos que podrían avanzar hacia la confirmación de esta hipótesis65. Entre estos datos se encuentran el desarrollo de un primer modelo animal de enfisema autoinmunitario66–68. Se ha correlacionado la presencia de autoanticuerpos antielastina69 y de otros autoantígenos70,71 con el grado de enfisema y se ha descrito que la inducción de autoanticuerpos contra las proteínas de la matriz pulmonar aumentan la respuesta inflamatoria inducida por el humo de tabaco en ratones previamente inmunizados con una mezcla de proteínas de la matriz extracelular pulmonar72.
Modelos de ensayos terapéuticos en la enfermedad pulmonar obstructiva crónicaLos tratamientos actuales tienen una eficacia limitada en la inhibición de la inflamación crónica, no revierten la patología de la EPOC y no modifican los factores que inician y conducen a la progresión de la enfermedad a largo plazo. Por lo tanto, existe una clara necesidad de nuevas terapias que puedan prevenir la inducción y la progresión de la EPOC, y los diseños de modelos animales que reflejen con exactitud la fisiopatología de la enfermedad siguen siendo esenciales para el desarrollo de nuevas terapias. Muchos de los fármacos en desarrollo clínico de la EPOC han sido identificados a partir de los trabajos realizados en modelos animales. Varios inhibidores de mediadores de inflamación están en desarrollo para el tratamiento de la EPOC. Pero hasta la fecha los inhibidores de LTB4, TNF-α, IL-1, IL-8 y EGF no han dado los resultados esperados73. Animales expuestos al humo de tabaco tratados con inhibidores sintéticos de la elastasa neutrofílica han puesto de manifiesto su potencial actividad antiinflamatoria74. Del mismo modo, la potencial utilidad terapéutica de los inhibidores de quinasas (p38 MAPK y PI3K) en la EPOC ha sido apoyada por los datos generados en modelos animales de inflamación pulmonar inducida por la exposición al humo de tabaco75. La enzima antioxidante Gpx-1 protege contra la inflamación pulmonar y el enfisema inducidos por el humo del cigarrillo en ratones76, y el Gpx mimético también reduce la inflamación pulmonar cuando se administra tanto profiláctica como terapéuticamente76,77. En otros estudios la deleción del gen de respuesta antioxidante Nrf2 conduce a aumentos en la inflamación pulmonar y enfisema en los ratones expuestos al humo del cigarrillo78, y un activador de Nrf2 se encuentra actualmente en ensayos clínicos de la EPOC73.
El descubrimiento de nuevos fármacos que puedan reducir la tasa de destrucción pulmonar y la limitación al flujo aéreo, e incluso detener o revertir los procesos subyacentes, sigue siendo una deuda pendiente del tratamiento de esta enfermedad. En este sentido, existen algunos datos acerca de que el ácido retinoico aminora notablemente el enfisema inducido en ratas por la administración de elastasa79, lo que ha suscitado un interés considerable en los retinoides80 y otros factores de crecimiento como potenciales agentes terapéuticos81, aunque por ahora no existe confirmación acerca de su potencial aplicabilidad. Datos publicados por nuestro grupo muestran que el factor de crecimiento de hígado (LGF) presenta una actividad antifibrótica pulmonar y es capaz de mejorar la función pulmonar y de revertir, al menos parcialmente, el incremento de las proteínas de matriz producido tras la instilación con CdCl2 en un modelo de fibrosis en ratas82.
En conclusión, el soporte de información que puede obtenerse a partir de los modelos animales de la EPOC sigue estando vigente y proporciona una buena herramienta para progresar en el conocimiento de los aspectos patogénicos de la enfermedad y de potenciales ensayos terapéuticos. La heterogeneidad que existe en la propia enfermedad también puede reflejarse en los modelos animales, que se desarrollan gracias al uso de diferentes combinaciones de agentes de inducción o de intensidades de los mismos. Por eso es importante elegir el modelo en función de la pregunta de investigación que cada caso requiera, sea de patogenia, de diagnóstico, de tratamiento o combinada.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.







