



Introducción
Las enfermedades pulmonares intersticiales difusas (EPID) constituyen un grupo heterogéneo de procesos que presentan manifestaciones clínicas, radiológicas y funcionales respiratorias comunes, y que afectan a las estructuras alveolointersticiales del pulmón y, a menudo, a las pequeñas vías aéreas y la vasculatura pulmonar.
Su clasificación se ha modificado recientemente, tras el documento de consenso elaborado por la American Thoracic Society y la European Respiratory Society, para distinguir 3 grupos de EPID: las neumonías intersticiales idiopáticas (cuya caracterización y definición histológica ha suscitado gran interés en los últimos años), las EPID de causa conocida o asociadas a entidades bien definidas (p. ej., enfermedades del colágeno), y un tercer grupo que incluye enfermedades granulomatosas y otros procesos que, aunque idiopáticos, presentan una clínica e histología bien definidas1-3.
La fibrosis pulmonar idiopática (FPI) es la más frecuente de las EPID4, con una prevalencia estimada de 20/100.000 habitantes en varones y de 13/100.000 en mujeres. Su etiología es desconocida y, dentro de las neumonías intersticiales idiopáticas, es la de peor pronóstico. Histopatológicamente, la FPI se caracteriza por la presencia de neumonía intersticial usual en la biopsia pulmonar de un paciente en el que se han excluido otras causas conocidas de EPID, y que presenta alteraciones clinicofuncionales compatibles y hallazgos característicos en la tomografía computarizada de alta resolución. El patrón anatomopatológico de la neumonía intersticial usual es heterogéneo, con áreas de fibrosis pulmonar que coexisten con focos de proliferación fibroblástica. Los focos de fibroblastos se localizan en el intersticio pulmonar, habitualmente en la frontera entre el tejido sano y el tejido pulmonar fibroso, y se caracterizan por la proliferación de fibroblastos y miofibroblastos, disminución de la apoptosis e hiperrespuesta a citocinas fibrogénicas. El componente inflamatorio de la neumonía intersticial usual es poco relevante5,6.
Clásicamente, la teoría fisiopatológica aceptada para la FPI sostenía que un agente causal desconocido produciría una inflamación crónica del tejido pulmonar que conduciría con el tiempo al desarrollo de fibrosis. Sin embargo, el uso de fármacos antiinflamatorios e inmunodepresores no ha demostrado ser eficaz en el tratamiento de la enfermedad. Actualmente se considera que el acontecimiento principal en el desarrollo de la FPI es la lesión celular del epitelio alveolar, que estimula el desarrollo de fibrosis, y que la inflamación representa un proceso secundario6.
Una característica esencial de la FPI es la proliferación de fibroblastos y la acumulación anormal de moléculas de la matriz extracelular, especialmente fibras colágenas. Los focos de fibroblastos, distribuidos ampliamente en el parénquima pulmonar, se comportan como pequeñas áreas de lesiones pulmonares agudas, adonde migran los fibroblastos, proliferan y contribuyen a la acumulación de moléculas de la matriz extracelular que dañan los alveolos. Así, como consecuencia de los depósitos intersticiales e intraluminales de tejido conectivo, se produce un "remodelado" de la arquitectura del pulmón.
Por otra parte, los fibroblastos no son una población homogénea. En la última década ha surgido el concepto de "diversidad fenotípica" al observarse, por ejemplo, que los fibroblastos en la FPI muestran un fenotipo secretorio profibrótico, con menor tasa de crecimiento. Los miofibroblastos son la única población de fibroblastos que expresan características de diferenciación de músculo liso. Los fibroblastos positivos para la α-actina de músculo liso aumentan en el tejido pulmonar de los pacientes con FPI y constituyen el principal componente de los focos fibroblásticos. Además, estas células adquieren un fenotipo "agresivo" y parece que son las principales responsables de la acumulación de colágeno. Asimismo, el remodelado que se observa en la matriz extracelular anormal de los pulmones de pacientes con FPI es, al menos en parte, debido a un desequilibrio entre algunos componentes de la familia de las metaloproteinasas de la matriz, como la colagenasa 1 (metaloproteinasa 1) o las gelatinasas A y B (metaloproteinasas 2 y 9, respectivamente), e inhibidores tisulares de metaloproteinasas. Los fibroblastos parecen, pues, desempeñar un papel esencial en este proceso de remodelado aberrante, ya que son responsables de la producción exagerada de diversos componentes de la matriz extracelular6-8.
En el desarrollo de la fibrosis pulmonar están implicadas muchas citocinas, como la interleucina 1, el factor de crecimiento derivado de las plaquetas, el factor de necrosis tumoral alfa (TNF-α) y, muy especialmente, el factor beta de transformación del crecimiento (TGF-β), considerado, quizá, el mediador fibrogénico más importante9 (tabla I). El TGF-β es producido por células inflamatorias y células epiteliales dañadas. Entre sus acciones destacan el estímulo de la síntesis de colágeno y de otros componentes de la matriz extracelular, así como la activación de inhibidores de proteasas, que degradan la matriz extracelular, y del inhibidor de la activación del plasminógeno. El TGF-β se secreta de forma inactiva, unido en su extremo aminoterminal al péptido asociado a la latencia. Posteriormente, la integrina αVβ6 se une a este péptido y activa el TGF-β10. Se han llevado a cabo experimentos en los que se ha demostrado que los inhibidores del TGF-β disminuyen la fibrosis pulmonar inducida por bleomicina.

Por último, en los últimos años se ha destacado el papel relevante que el estrés oxidativo, los radicales libres y la ruptura del equilibrio oxidantes/antioxidantes pueden desempeñar en el desarrollo la de fibrosis pulmonar.
Nos faltan piezas por encajar dentro de este complejo escenario, pero estas y otras investigaciones abren la puerta a nuevas posibilidades terapéuticas11,12.
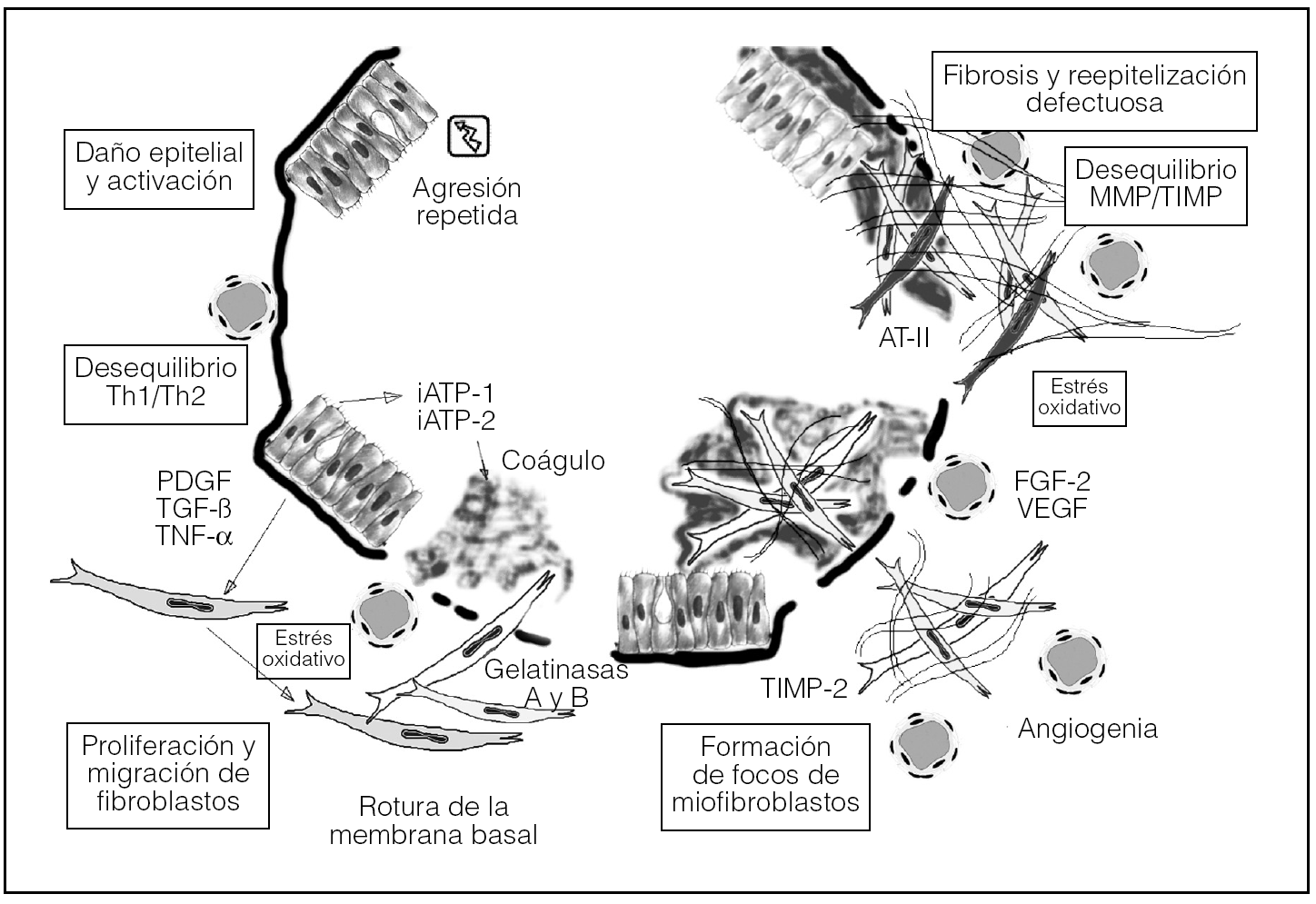
En la figura 1 se esquematizan los componentes esenciales implicados en la patogenia de la FPI.

Fig. 1. Esquema patogénico secuencial de la fibrosis pulmonar idiopática, una enfermedad multicomponente (hipótesis de Selman modificada). AT-II: angiotensina II; FGF-2: factor 2 de crecimiento fibroblástico; iATP-1 e iATP-2: inhibidores 1 y 2 del activador tisular del plasminógeno; PDGF: factor de crecimiento derivado de plaquetas; MMP: metaloproteinasas; TGF-β: factor β transformador de crecimiento; Th: linfocitos T helper; TIMP: inhibidores tisulares de metaloproteinasas; TNF-α: factor de necrosis tumoral alfa; VEGF: factor de crecimiento del endotelio vascular.
Tratamiento clásico de la fibrosis pulmonar idiopática
Durante décadas el tratamiento de la FPI ha ido encaminado a combatir la inflamación mediante el uso de glucocorticoides, asociados o no a inmunodepresores como la azatioprina y ciclofosfamida. Por desgracia, ninguno de estos fármacos ha demostrado ser realmente útil a la hora de mejorar el pronóstico infausto de la mayoría de los pacientes.
La mayor parte de los estudios que han valorado la eficacia de estos fármacos son controvertidos. Han incluido, con toda probabilidad, a pacientes con otros tipos de neumopatías intersticiales menos radicales (neumonía intersticial no específica, neumonía intersticial descamativa y neumopatía intersticial asociada a enfermedades del colágeno).
De las estrategias terapéuticas, la única que ha demostrado ser útil para mejorar ligeramente la supervivencia es la asociación de glucocorticoides y azatioprina. De los escasos trabajos publicados, el más consistente es el de Raghu et al13, quienes realizaron un estudio prospectivo, aleatorio, doble ciego y controlado para comparar la asociación de azatioprina y prednisona frente al tratamiento con prednisona. Reclutaron a pacientes con neumonía intersticial usual diagnosticada por biopsia pulmonar abierta, excluyendo a aquellos con otras formas de EPID. Aunque la muestra fue pequeña, se logró demostrar un aumento moderado de la supervivencia en el grupo con azatioprina, particularmente cuando se controlaba por edad.
La normativa de la Sociedad Española de Neumología y Cirugía Torácica y los paneles de expertos de la American Thoracic Society y la European Respiratory Society recomiendan la administración de glucocorticoides asociados a ciclofosfamida o azatioprina (tabla II)1-3; esta última se utiliza más debido a que produce menos efectos secundarios. La duración del tratamiento es variable, pero se aconseja mantener la pauta inicial al menos durante 6 meses. La colchicina, fármaco con propiedades antifibróticas, puede representar una alternativa en los pacientes con mala tolerancia a glucocorticoides e inmunodepresores. El estudio más destacado en este campo fue el que llevaron a cabo Douglas et al14 en 1998. Reclutaron a 26 pacientes diagnosticados de FPI; a 12 de ellos les administraron prednisona a dosis altas (60 mg/día como mínimo) y, al resto, colchicina (0,6-1,2 mg/día). No se observaron diferencias estadísticamente significativas en cuanto a supervivencia y función pulmonar en ambos grupos de pacientes. Sin embargo, la colchicina resultó ser más segura y mejor tolerada.

Otros fármacos como la ribavirina, la d-penicilamina, el clorambucil y el metotrexato sólo se han utilizado en casos aislados, y su verdadera eficacia se desconoce.
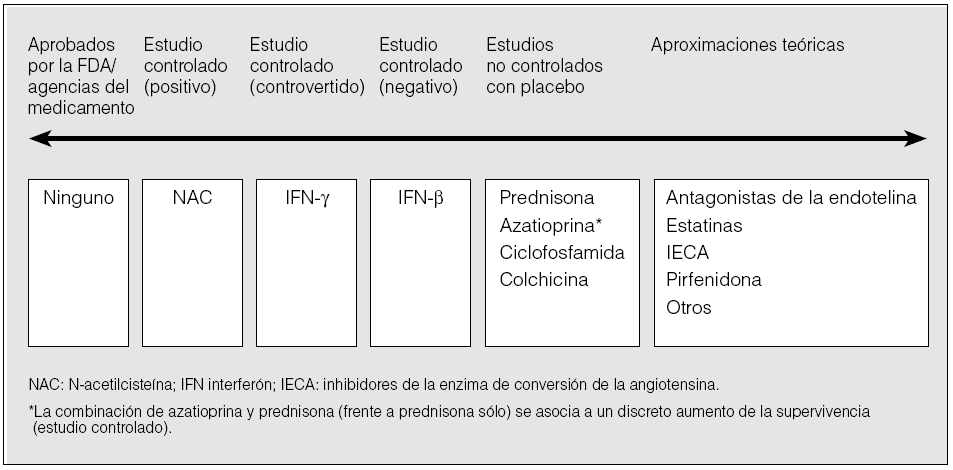
En el momento actual, constituye un auténtico reto intelectual y científico la necesidad de encontrar alternativas terapéuticas más selectivas y eficaces (fig. 2). En este contexto, resultan especialmente atractivas determinadas líneas de trabajo que tratan de definir el papel real de los antioxidantes, el interferón gamma (IFN-γ), los fármacos anti-TNF-α y, particularmente, el bloqueo de TGF-β en el tratamiento de la FPI.

Fig. 2. Evidencias en las que se sustenta el uso de los diferentes agentes terapéuticos en la fibrosis pulmonar idiopática. FDA: Food and Drug Administration; NAC: N-acetilcisteína; IFN: interferón; IECA: inhibidores de la enzima de conversión de la angiotensina.
N-acetilcisteína (NAC)
Existen evidencias que constatan la presencia de un desequilibrio entre oxidantes y antioxidantes como parte de la patogenia de la FPI. En efecto, la FPI se caracteriza por un estrés oxidativo excesivo en el tracto respiratorio inferior. Las concentraciones de glutatión (el mayor componente del sistema de defensa antioxidante del pulmón humano, que, en condiciones normales, protege al tracto respiratorio inferior del estrés oxidativo) se encuentran reducidas de forma importante en la superficie epitelial alveolar de los pacientes con FPI.
En las últimas 2 décadas, la NAC se ha utilizado ampliamente en el campo de la medicina respiratoria como agente mucolítico. Más recientemente se ha establecido su poder antioxidante. En vivo, la NAC ejerce su función antioxidante a través de su principal metabolito, la cisteína, el mayor precursor en la biosíntesis del glutatión. Diversos estudios a corto plazo han planteado que la NAC, por vía oral o intravenosa, es capaz de aumentar las concentraciones reducidas de glutatión en el fluido epitelial pulmonar de los pacientes con fibrosis pulmonar15,16. Así, por ejemplo, en el estudio de Behr et al17 se administró NAC a dosis altas (600 mg 3 veces al día) durante 6 semanas a 18 pacientes con FPI que estaban recibiendo tratamiento inmunodepresor. Se realizaron pruebas de función respiratoria y lavado broncoalveolar antes y después de la administración del tratamiento. Demostraron un aumento significativo de los valores de glutatión y una reducción, también significativa, de las concentraciones de sulfóxido de metionina (indicador del estrés oxidativo) en el lavado broncoalveolar. Estos cambios bioquímicos se acompañaron de una mejoría significativa de la función pulmonar en pacientes con FPI que habían recibido tratamiento inmunodepresor durante al menos 6 meses. Se concluyó, por tanto, que la NAC debería añadirse al tratamiento clásico en pacientes con FPI.
Dada su elevada seguridad y tolerancia, además de las teóricas ventajas basadas en su implicación patogénica, la NAC ha despertado un interés creciente en el tratamiento combinado de la FPI. De hecho, distintos grupos de Bélgica, Francia, Alemania, Italia, Países Bajos, Reino Unido y España han llevado a cabo el estudio IFIGENIA (Idiopathic Pulmonary Fibrosis International Group Exploring NAC I Annual), sobre los efectos de la NAC a dosis altas en la FPI. Se trata de un estudio multicéntrico, aleatorio, doble ciego y controlado con placebo cuyo objetivo principal es conocer si el tratamiento de la FPI con NAC más prednisona en asociación con azatioprina tiene un mejor efecto en la función pulmonar (variables principales: capacidad vital y capacidad de difusión de monóxido de carbono), en la radiología y en la situación clínica que el uso de placebo más prednisona y azatioprina después de 6 y 12 meses. Se cumplieron las expectativas. En el congreso de la European Respiratory Society celebrado recientemente en Glasgow (septiembre de 2004), se presentaron los resultados del estudio IFIGENIA, que ponen de manifiesto que la adición de NAC oral a una dosis alta (600 mg 3 veces al día) al tratamiento clásico tiene un efecto beneficioso a medio plazo sobre la función pulmonar de pacientes con FPI, con un excelente perfil de cumplimentación y seguridad18.
Fármacos antifactor de necrosis tumoral alfa
Parece esencial el papel que cumplen determinadas citocinas profibróticas, como el TGF-β y el TNF-α, en el proceso que conduce a la formación de focos fibroblásticos y la subsiguiente fibrosis pulmonar. La liberación de citocinas proinflamatorias con capacidad fibrogenética en las fases inciales de la FPI hace que un objetivo lógico del tratamiento sea una actuación directa en este nivel.
El TNF-α es liberado por los macrófagos alveolares y los linfocitos T. Actúa reclutando y liberando células inflamatorias, promueve la inflamación crónica e induce la proliferación de los fibroblastos. Varios grupos han demostrado que el TNF-α se sintetiza y secreta en exceso en la FPI, tanto en modelos animales como en humanos. Trabajos como los de Piquet et al19,20 recalcan la importancia de los fármacos anti-TNF-α como agentes bloqueadores de la fibrogenia. Estos autores demuestran que la fibrosis pulmonar inducida por bleomicina en ratones puede detenerse con la administración de anti-TNF-α o su receptor.
No obstante, la mayoría de los estudios que refuerzan la importancia de estos fármacos proviene del campo de la reumatología21. De hecho, se están utilizando fármacos anti-TNF-α en la fibrosis pulmonar asociada a artritis reumatoide. Hasta la fecha, no existen datos controlados en la FPI, si bien van a iniciarse ensayos clínicos en fase III en esta enfermedad.
Interferón gamma-1b
El IFN-γ es una citocina endógena que tiene efectos antifibróticos, antiinfecciosos, antiproliferativos e inmunomoduladores. En la FPI existe un desequilibrio entre las citocinas producidas por los linfocitos T helper tipo 2 y las producidas por los linfocitos T helper 1 a favor de las primeras. El IFN-γ altera este cociente a favor de los linfocitos T helper 122.
En estudios in vitro y con modelos animales se ha demostrado que el IFN-γ inhibe la proliferación de los fibroblastos, el depósito de matriz extracelular y la síntesis de colágeno, además de disminuir la expresión de TGF-β. Se han llevado a cabo varios estudios para analizar la eficacia del IFN-γ en pacientes con FPI. Ziesche et al23 realizaron un ensayo clínico en fase II con 18 pacientes que no respondían al tratamiento convencional y obtuvieron resultados muy alentadores. Los pacientes tratados con IFN-γ más prednisolona mejoraban su función pulmonar respecto a los tratados exclusivamente con prednisolona. No obstante, se trataba de una cohorte muy pequeña y, al revisar el estudio, no todos los pacientes cumplían realmente los criterios diagnósticos de FPI. Posteriormente, Prasse et al24 realizaron un ensayo similar con 5 pacientes y no pudieron demostrar los resultados del estudio anterior. Ambos trabajos apuntaban, en todo caso, a que probablemente el IFN-γ es más eficaz en las primeras fases de la enfermedad que en estadios más avanzados.
No obstante, las expectativas generadas por el IFN-γ no se han visto todavía satisfechas. Recientemente Raghu et al25 han llevado a cabo un ensayo clínico multicéntrico, aleatorio, doble ciego y controlado con placebo en el que han incluido a 330 pacientes diagnosticados de FPI, sin respuesta al tratamiento con antiinflamatorios e inmunodepresores. Durante un año los pacientes han recibido tratamiento con IFN-γ subcutáneo o placebo. Los autores no encontraron diferencias significativas en cuanto a la progresión de la enfermedad o la supervivencia, y tampoco en la función pulmonar, el intercambio de gases en reposo o la calidad de vida. No obstante, observaron una tendencia a mejorar la supervivencia en los pacientes tratados con IFN-γ en relación con el grupo control, y ésta era mayor en aquellos que presentaban buena adherencia al tratamiento. Son, pues, necesarios estudios más amplios y de mayor duración para saber si el IFN-γ aumenta la supervivencia en los pacientes con FPI. En este sentido, se ha iniciado recientemente el estudio INSPIRE. Se trata de un ensayo en fase III, aleatorio (2:1), doble ciego y controlado con placebo que analiza la eficacia y seguridad del IFN-γ-1b recombinante subcutáneo durante 2 años en una muestra amplia de pacientes con FPI.
Pirfenidona
El efecto antifibrótico de la pirfenidona ha quedado demostrado en diversos estudios tanto in vitro como en modelos animales. In vitro, la pirfenidona inhibe la síntesis de colágeno estimulada por el TGF-β, disminuye la formación de matriz extracelular y reduce la producción de citocinas fibrogénicas como el TNF-γ, el TGF-β y el factor de crecimiento derivado de las plaquetas. En animales con fibrosis pulmonar inducida por bleomicina se ha visto que la pirfenidona disminuye el desarrollo de ésta. En 1999 Raghu et al26 realizaron un ensayo clínico en fase II en el que incluyeron a 54 pacientes en estadio avanzado de FPI que presentaban deterioro a pesar del tratamiento convencional, o bien intolerancia a éste. En la mayoría de los pacientes, tras el tratamiento con pirfenidona se observó una estabilización de la enfermedad y, en algunos, una mejoría de la oxigenación. El fármaco se toleró relativamente bien y permitió disminuir la dosis de corticoides e inmunodepresores en la mayoría de los pacientes sin que esto supusiera un deterioro de la función pulmonar. Los resultados de este estudio, si bien alentadores, no son concluyentes, por lo que son necesarios nuevos ensayos aleatorios que determinen el efecto real de la pirfenidona en la supervivencia.
Más recientemente, un estudio japonés realizado por Nagai et al27 analiza el efecto de la pirfenidona en pacientes con FPI y enfermedad intersticial asociada a esclerosis sistémica. El tratamiento con pirfenidona no demostró un efecto terapéutico sobre la supervivencia, pero durante el año que duró el estudio no se observó un deterioro significativo radiológico ni en la presión parcial de oxígeno en sangre arterial.
Lovastatina
Algunos autores han planteado como hipótesis la inducción de apoptosis en las células fibroblásticas para frenar la progresión hacia la fibrosis.
La lovastatina es un fármaco muy utilizado en el tratamiento de la hipercolesterolemia, ya que inhibe la enzima 3-hidroxi-3-metilglutaril coenzima A reductasa. Además, es conocida su función en el mantenimiento de la homeostasis, así como en la proliferación y supervivencia celulares. Tan et al28 han demostrado que determinadas concentraciones de lovastatina son capaces de inducir apoptosis de fibroblastos pulmonares humanos in vitro. En vivo, trabajando con modelos de fibroproliferación en cerdos se observó una reducción en la formación de tejido de granulación y la inducción de apoptosis fibroblástica.
Inhibidores de la enzima de conversión de la angiotensina
Es un hecho demostrado la presencia de componentes del sistema renina-angiotensina y la elevación de la enzima de conversión de la angiotensina (ECA), en el lavado broncoalveolar o en el suero, en numerosas enfermedades fibrosantes pulmonares, tales como asbestosis, silicosis, sarcoidosis y FPI29.
La elevación de la ECA en el lavado broncoalveolar puede inducir la elevación de angiotensina II, lo que hace pensar que ambas moléculas puedan tener algún papel en el desarrollo de la fibrosis pulmonar30. El tratamiento con inhibidores de la ECA ha sido muy estudiado en enfermedades del corazón y riñón, en relación con hipertrofia, hiperplasia y fibrosis de estos tejidos. Más recientemente, nuevos estudios se han centrado en el tratamiento o prevención de la enfermedad hepática y pulmonar.
Marshall et al31 observaron que la angiotensina II tiene un efecto mitogénico sobre los fibroblastos pulmonares. Vieron que dicho efecto estaba mediado por el receptor tipo 1 de la angiotensina II, de manera que bloqueando dicho receptor con fármacos específicos como el losartán o el irbesartán no se producía el proceso de fibrosis estimulado por la angiotensina II, mientras que el proceso continuaba su curso si lo que se bloqueaba era el receptor tipo 2.
Otros investigadores han encontrado una clara relación entre la angiotensina II y el TGF-β1. Así, la estimulación con angiotensina II o la inhibición de sus receptores produce un aumento o descenso, respectivamente, de las concentraciones de TGF-β, y estas concentraciones parecen relacionarse directamente con un aumento de la fibrosis o con una protección frente a ella.
Al menos en teoría, podemos considerar, como posible tratamiento futuro, el empleo de antagonistas específicos de los receptores tipo 1 de la angiotensina II (losartán e irbesartán), con el fin de romper la cadena de interacciones moleculares y detener o dificultar el desarrollo de la fibrosis pulmonar32.
Hipertensión pulmonar y enfermedades pulmonares intersticiales
Las EPID se asocian frecuentemente al desarrollo de hipertensión pulmonar. Su patogenia se ha visto relacionada no sólo con la alteración del lecho vascular sino con la presencia de un potente vasoconstrictor pulmonar endógeno, la endotelina 1 (ET-1). Los pacientes con hipertensión pulmonar, tanto primaria como secundaria, presentan valores arteriales elevados de ET-1, y estos valores son aún mayores en pacientes con hipertensión pulmonar asociada a otras enfermedades. Recientes estudios in vitro demuestran que la ET-1, además de su efecto vasoconstrictor, estimula la proliferación de fibroblastos y miofibroblastos y la síntesis de proteínas de la matriz extracelular por éstos. Por tanto, en teoría el bloqueo de la ET-1 disminuiría la presión en los vasos pulmonares y reduciría los procesos inflamatorios y proliferativos asociados en las EPID33,34.
El bosentán es un antagonista oral de ambos receptores de la ET-1: ETA y ETB. Rubin et al35 estudiaron el efecto del bosentán en 213 pacientes con hipertensión pulmonar primaria o asociada a conectivopatías. Observaron que la administración de 125 mg de bosentán 2 veces al día mejoraba significativamente la capacidad de ejercicio y retrasaba el empeoramiento clínico en los pacientes con hipertensión pulmonar grave. Los pacientes presentaban una mejoría de su disnea y de su clase funcional. El ensayo, sin embargo, no tenía en cuenta la hipertensión pulmonar asociada a otros procesos ni el efecto del bosentán en la supervivencia, por lo que no obvia la necesidad de estudios complementarios. Para definir el papel del bosentán en pacientes con EPID se están llevando a cabo 2 ensayos clínicos internacionales que está previsto finalicen a mediados de 2005: el estudio BUILD-1 (en sujetos con FPI) y el estudio BUILD-2 (en enfermos con EPID asociada a esclerosis sistémica progresiva).
El sildenafilo es un inhibidor de la fosfodiesterasa que se administra oralmente, prolonga el efecto vasodilatador del óxido nítrico y mejora el intercambio de gases en pacientes con hipertensión pulmonar. Se ha comparado su efecto con el del epoprostenol en pacientes con hipertensión pulmonar secundaria a fibrosis pulmonar y se ha visto que ambos tratamientos disminuyen las resistencias vasculares pulmonares. En este ensayo, el sildenafilo se asociaba con un descenso en la fracción del shunt y, por tanto, con una mejoría en la saturación arterial de oxígeno, mientras que el epoprostenol aumentaba el shunt intrapulmonar y disminuía la oxigenación arterial. Aunque estos resultados son prometedores, son necesarios ensayos clínicos en fase III en pacientes con FPI36.
Trasplante pulmonar en la fibrosis pulmonar idiopática
El trasplante pulmonar es la última opción terapéutica para pacientes seleccionados en la fase final de la enfermedad. El trasplante pulmonar puede mejorar la calidad de vida y aumentar la supervivencia. De acuerdo con los recientes datos internacionales, la FPI representa el 2,9% de los trasplantes de corazón-pulmón, el 19,5% de los trasplantes unipulmonares y el 7,1% de los trasplantes bipulmonares llevados a cabo en población adulta37.
De los diferentes procedimientos disponibles para el trasplante pulmonar (trasplante unipulmonar, trasplante bipulmonar, trasplante corazón-pulmón y trasplante lobar), el trasplante unipulmonar es el procedimiento de elección en los pacientes afectados de FPI. Los candidatos a trasplante deben cumplir los requisitos generales de cualquier trasplante pulmonar y no presentar contraindicaciones.
Otras vías de investigación
Dada la complejidad de la patogenia de la FPI, es posible desarrollar distintos fármacos que controlen su progresión y que actúen por vías moleculares distintas. Por ejemplo, el tratamiento combinado de taurina y niacina en modelos animales de fibrosis pulmonar inducida por bleomicina tiene efectos antifibróticos al bloquear la sobreexpresión de TGF-β a nivel transcripcional. En otros ensayos experimentales se ha observado que los anticuerpos anti-TGF-β reducen de manera significativa la fibrosis renal y pulmonar. Las metaloproteinasas son colagenasas que presentan valores reducidos en pacientes con FPI. Los anticuerpos anti-TGF-β antagonizan los efectos de los inhibidores tisulares de las metaloprotasas, por lo que aumentan la actividad colagenolítica. Otra molécula con efecto antifibrótico es la decorina, una molécula humana que podría administrarse en forma de aerosol y no produciría reacciones adversas inmunológicas como los anticuerpos anti-TGF-β. Como ya se ha expuesto anteriormente, el TGF-β tiene un papel predominante en el desarrollo de la FPI y es activado por la integrina αVβ6, por lo que sería lógico pensar que el bloqueo de esta activación disminuiría la fibrosis. Por último, futuras estrategias incluyen el bloqueo de elementos de transducción de señalización celular y estrategias específicas de bloqueo de transfección genética38-40.
Mientras tanto, en el momento actual, y desde un punto de vista conceptual, probablemente el tratamiento más adecuado de la FPI (enfermedad multicomponente) sea un "tratamiento combinado", que incluya un fármaco antifibrótico, un inmunodepresor y un antioxidante (fig. 3). Es preciso seguir investigando y que clínicos y básicos aunemos esfuerzos con un objetivo común. Quizá así consigamos que la FPI sea realmente una "enfermedad tratable".

Fig. 3. Fibrosis pulmonar idiopática: nuevos esquemas terapéuticos en una "enfermedad multicomponente". IECA: inhibidores de la enzima de conversión de la angiotensina; IFN: interferón; TGF: factor transformador de crecimiento; TNF: factor de necrosis tumoral.







