








Children's diffuse lung disease, also known as children's Interstitial Lung Diseases (chILD), are a heterogeneous group of rare diseases with relevant morbidity and mortality, which diagnosis and classification are very complex. Epidemiological data are scarce. The aim of this study was to analyse incidence and prevalence of chILD in Spain.
MethodsMulticentre observational prospective study in patients from 0 to 18 years of age with chILD to analyse its incidence and prevalence in Spain, based on data reported in 2018 and 2019.
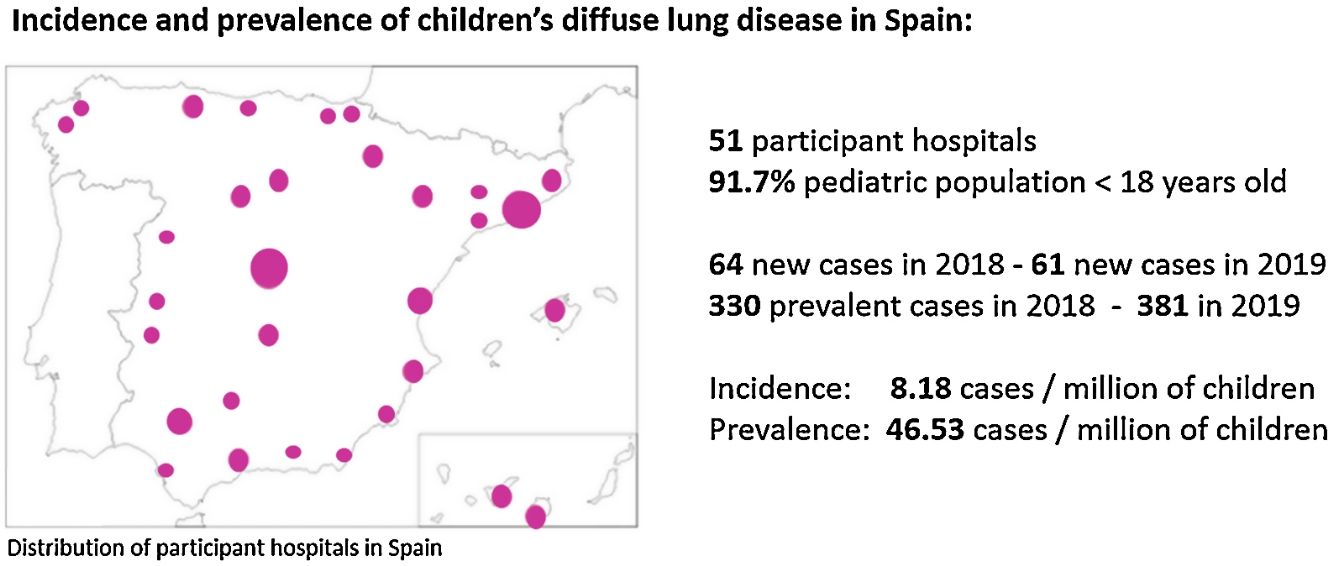
ResultsA total of 381 cases with chILD were notified from 51 paediatric pulmonology units all over Spain, covering the 91.7% of the paediatric population. The average incidence of chILD was 8.18 (CI 95% 6.28–10.48) new cases/million of children per year. The average prevalence of chILD was 46.53 (CI 95% 41.81–51.62) cases/million of children. The age group with the highest prevalence were children under 1 year of age. Different types of disorders were seen in children 2–18 years of age compared with children 0–2 years of age. Most frequent cases were: primary pulmonary interstitial glycogenosis in neonates (17/65), neuroendocrine cell hyperplasia of infancy in infants from 1 to 12 months (44/144), idiopathic pulmonary haemosiderosis in children from 1 to 5 years old (13/74), hypersensitivity pneumonitis in children from 5 to 10 years old (9/51), and scleroderma in older than 10 years old (8/47).
ConclusionsWe found a higher incidence and prevalence of chILD than previously described probably due to greater understanding and increased clinician awareness of these rare diseases.
Las neumopatías intersticiales pediátricas, también conocidas con el acrónimo chILD (del inglés children's Interstitial Lung Diseases), es un grupo heterogéneo de enfermedades raras con morbimortalidad relevante, cuyo diagnóstico y clasificación son complejos. Los estudios epidemiológicos son escasos. El objetivo de este trabajo fue analizar la incidencia y la prevalencia de chILD en España.
MétodosEstudio prospectivo observacional multicéntrico en pacientes de 0 a 18 años afectos de chILD para analizar la incidencia y la prevalencia en España, a partir de datos recogidos en 2018 y 2019.
ResultadosSe recogieron 381 casos de chILD entre 51 unidades de neumología pediátrica de toda España, que cubrían el 91,7% de la población pediátrica. La incidencia promedio fue 8,18 (IC 95%: 6,28-10,48) casos nuevos/millón de niños por año. La prevalencia promedio fue de 46,53 (IC 95%: 41,81-51,62) casos/millón de niños. El grupo de edad con mayor prevalencia fue el de niños menores de un año. Se observaron diferentes entidades en niños de 2 a 18 años en comparación con niños de 0 a 2 años. Los diagnósticos más frecuentes fueron: glucogenosis intersticial pulmonar primaria en neonatos (17/65), hiperplasia de células neuroendocrinas en lactantes de uno a 12 meses (44/144), hemosiderosis pulmonar idiopática en niños de uno a 5 años (13/74), neumonía por hipersensibilidad en niños de 5 a 10 años (9/51) y esclerodermia en mayores de 10 años (8/47).
ConclusionesEncontramos una mayor incidencia y prevalencia de chILD que las descritas previamente, probablemente debido a un mayor conocimiento y detección de estas enfermedades raras.
Paediatric diffuse lung diseases are a heterogeneous group of rare diseases with significant morbidity and mortality which manifest from newborns to adolescents, characterised by respiratory impairment and diffuse abnormalities on imaging studies.1 These groups of diffuse lung diseases are known by the acronym chILD (Children's Interstitial Lung Diseases), proposed by the North American group ChILD Research Network (chILDRN),2 although many of these disorders affect the lung structure beyond the interstitium.
Diagnosis and categorisation of chILD is complex, due to their diversity and very low frequency. Collaborative efforts have led to a classification system more appropriate for infants and children with chILD, based on histological, clinical and imaging findings. Two age groups are distinguished: disorders of children under 2 years old, which are very different from adults, and disorders seen in older children (2–18 years old), who present a more similar spectrum of diseases compared to adults. Nevertheless, some particularities due to the influence of lung development and the early onset of the disease must be considered.3–5 Since the initial classification, categorisation of chILD has evolved because new entities have been described in newborns, infants and older children, and new disease causing mutations have been identified.6–8
Some studies have analysed chILD incidence and prevalence in different populations, being several times lower than in adults.9 First data published estimated a prevalence of 3.6 cases per million of children in United Kingdom and Ireland.10 Incidence has been estimated ranging from 1.32 new cases per million of children per year in Germany11 to 108–162 cases per million of children under 14 years old in a study carried out in Denmark in all range of ages.12 More recently, the Australasian Registry Network for Orphan Lung Disease published their experience over the last decade, estimating the prevalence at 1.5 (0.8–2.1) cases per million for children.13 Nevertheless, these reported measures of chILD frequency might be underestimated, due to different diagnostic criteria and disease definitions, restricted access to genetic testing, under-recognition and under-reporting. Moreover, new entities have been identified since then, and chILD detection has been increasing as its understanding has spread over together with greater clinician awareness and improved use of diagnostic tools.1,14,15 National and international registries of chILD cases have contributed to improve our knowledge, but since their implementation is not exhaustive, they do not allow us to know well the incidence and prevalence of these diseases. It is unknown whether incidence and prevalence are the same in different populations. Most of the studies have been carried out in the Anglo-Saxon or Central European area, and there are no data from the Mediterranean basin. On the other hand, advances in identification of new diseases in this group may suggest that they are more frequent than has been established so far.13,16–18
With this background, we aimed to calculate incidence and prevalence of chILD in a large paediatric population of a Mediterranean country, based on data from most of the paediatric pulmonology units in Spain.
MethodsThis is a multicentre observational prospective study in patients from 0 to 18 years of age with chILD, to analyse their incidence and prevalence. Paediatric pulmonology units all over Spain were invited to participate. Those who agreed to participate received monthly email surveys from 1st January 2018 to 31st December 2019, to identify new cases each month. Besides, two transversal studies were done in October 2018 and October 2019, when participants reported all prevalent patients that were being followed-up. Anonymised questionnaires were sent to collect data from new and prevalent cases, requesting information about specific diagnosis and the age at first symptoms. Small centres that shared their patients with referral centres were asked to give notice in all cases, so we could track them and avoid duplicates. In order to minimise under-diagnosis, all participants were given case definitions and the list of diseases used for categorisation. This list was the same we subsequently used to analyse the results. In addition, there was a continuous and permanent contact with all the centres in order to collect all missing data. The study was approved by the Ethics Committee of Hospital Universitari Vall d’Hebron (Barcelona, Spain), and by local ethics committees that required additional independent approval. Informed consent was required from parents and children if older than 12 years when indicating results of genetic studies.
Cases were defined based on chILD Syndrome criteria,19 when 3 of the following conditions were present, after excluding common conditions presenting similarly: (1) respiratory symptoms (cough, rapid and/or difficult breathing, exercise intolerance); (2) respiratory signs (tachypnoea, adventitious sounds, retractions, digital clubbing, failure to thrive, respiratory failure); (3) hypoxemia; (4) diffuse abnormalities on a chest radiograph or computed tomography (CT) scan. Final diagnosis was made based on clinical information, CT scan abnormalities and in some cases also based on lung biopsy or genetic studies.
Diagnostic classification (Supplemental Table 1) was based on the classification of the chILD Research Network (chILDRN)2 and the clinical practice guideline of the American Thoracic Society,3 expanded by Rice et al.,7 and with new diseases identified afterwards.20–26 Cases that could not be categorised in a specific diagnosis were reported as lung disease with unknown cause. Patients were classified by the specific diagnosis and their age group (<1 month old, 1 month–1 year old, 1–2 years old, 2–5 years old, 5–10 years old and >10 years old).
The following specific lung diseases were excluded from our analysis: chronic lung disease of the preterm infant (bronchopulmonary dysplasia), lung disease secondary to malignancy, transplantation and rejection, aspiration, infectious and postinfectious processes, as well as diseases that could be misdiagnosed as chILD, like veno-occlusive disease or pulmonary congestive changes related to cardiac dysfunction. In the case of “Disorders of the immunocompromised host” only non-infectious interstitial lung involvement related to the underlying disease was included. The category “Chronic neonatal lung disease (not prematurity-related)” refers to term infants with alveolar growth abnormalities.2
The study of incidence and prevalence was based on data from two consecutive years, 2018 and 2019, to enable better tracking and analysis. Incidence rate was calculated as the sum of new cases diagnosed from 1st January to 31st December each year, expressed as number of cases per million of children per year. Prevalence rate was calculated as the sum of all alive cases that were currently followed-up in October 2018 and October 2019, expressed as number of cases per million of children. The reference population considered to calculate incidence and prevalence was the sum of residents under 18 years of each region with a hospital participating in the study or covered by a referral hospital participating in the study. These data were obtained from census data of the National Statistics Institute of Spain (https://www.ine.es). Descriptive statistical analysis was performed to yield medians, ranges and 95% confidence interval (CI) for quantitative variables.
ResultsFifty-eight paediatric pulmonology units situated all over Spain were asked to participate, from which 51 accepted. That means an overall return of surveyed centres of 88% covering a paediatric population of 7,644,155 in 2018 and 7,636,093 in 2019, representing 91.7% of population up to 18 years old in Spain. It was not possible to retrieve information from some regions of Spain with a paediatric population of 692,239 in 2018 and 687,862 in 2019. On the whole, epidemiological and clinical data was provided for 381 patients.
In 2018, 64 new cases of chILD were reported, and 330 were identified in the transversal study, whereas in 2019, there were 61 new cases and 381 were identified in the transversal study. There were 3 cases loss to follow-up between 2018 and 2019. Seven incident cases diagnosed in November–December 2019 were not included in the October 2019 prevalence calculation.
The median number of prevalent cases reported by each participating centre was 2 (range 0–68). Participant centres, paediatric reference population of each area, and data collection each year of the study period are shown in Supplemental Table 2.
The average incidence of paediatric diffuse lung disease was 8.18 (CI 95% 6.28–10.48) new cases/million of children per year. The average prevalence of paediatric diffuse lung disease was 46.53 (CI 95% 41.81–51.62) cases/million of children. Incidence and prevalence per year of the study period is shown in Table 1. Prevalence data stratified by age of patients at initial symptoms is provided in Table 2.
Incidence and Prevalence Each Year of the Study Period.
| Period of Study | 2018 | 2019 | Two Year Perioda |
|---|---|---|---|
| New cases | 64 | 61 | 62.5 |
| Prevalent cases | 330 | 381 | 355.5 |
| Population of reference (<18 y) | 7,644,155 | 7,636,093 | 7,640,124 |
| Incidence per million of children (CI 95%) | 8.37 (6.45–10.69) | 7.98 (6.10–10.25) | 8.18 (6.28–10.48) |
| Prevalence per million of children (CI 95%) | 43.17 (38.63–48.08) | 49.89 (45.00–55.16) | 46.53 (41.81–51.62) |
Prevalence Data of the Transversal Study in October 2019, Stratified by Age of Patients at Initial Symptoms.
| Age Group | <1 y | 1–2 y | 2–5 y | 5–10 y | >10 y |
|---|---|---|---|---|---|
| Prevalent cases | 209 | 22 | 52 | 51 | 47 |
| Population of reference | 329,092 | 719,503 | 1,207,312 | 2,184,274 | 3,195,912 |
| Prevalence per million of children (CI 95%) | 635.08 (551.91–727.23) | 30.58 (19.16–46.30) | 43.07 (32.17–56.48) | 23.35 (17.38–30.70) | 14.70 (10.80–19.54) |
CI: confidence interval; y: years.
Regarding prevalent cases, the most frequent categories among Disorders more prevalent in infancy were: specific conditions of undefined aetiology of infancy (73), growth abnormalities (47) and surfactant dysfunction mutations (22); and the most frequent categories among disorders not specific to infancy were: disorders related to systemic disease processes (67), lung disease with unknown cause (37) and idiopathic pulmonary haemosiderosis (24). Frequency and classification of reported prevalent chILD cases with specific diagnosis are presented in Table 3.
Frequency and Classification of Prevalent chILD Cases.
| Disorders Identified | Number of Cases | ||||||
|---|---|---|---|---|---|---|---|
| All Ages | <1 m | 1 m–1 y | 1–2 y | 2–5 y | 5–10 y | >10 y | |
| Disorders more prevalent in infancy | |||||||
| Diffuse developmental disorders | |||||||
| Acinar dysplasia | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Congenital alveolar dysplasia | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Alveolar capillary dysplasia with misalignment of pulmonary veins | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Growth abnormalities | |||||||
| Chronic neonatal lung disease (not prematurity-related) | 11 | 4 | 7 | 0 | 0 | 0 | 0 |
| Structural pulmonary changes with chromosomal abnormalitiesa | 24 | 8 | 11 | 1 | 4 | 0 | 0 |
| Associated with congenital heart disease in chromosomally normal children | 12 | 2 | 5 | 1 | 0 | 2 | 2 |
| Specific conditions of undefined aetiology | |||||||
| Primary pulmonary interstitial glycogenosisb | 23 | 17 | 6 | 0 | 0 | 0 | 0 |
| Pulmonary interstitial glycogenosis associated with other disordersb | 5 | 4 | 1 | 0 | 0 | 0 | 0 |
| Neuroendocrine cell hyperplasia of infancy | 45 | 1 | 44 | 0 | 0 | 0 | 0 |
| Surfactant dysfunction mutations and related disorders | |||||||
| Surfactant protein B mutations (SFTPB; R) | 2 | 0 | 2 | 0 | 0 | 0 | 0 |
| Surfactant protein C mutations (SFTPC; D) | 9 | 2 | 6 | 1 | 0 | 0 | 0 |
| ABCA3 mutations (ABCA3 bi-allelic mutations) | 5 | 1 | 4 | 0 | 0 | 0 | 0 |
| Brain–lung–thyroid syndrome (NKX2-1/TTF-1 mutations; D)c | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| Lysinuric protein intolerance (SLC7A7; R)c | 3 | 0 | 0 | 2 | 0 | 1 | 0 |
| Surfactant protein A mutations (SFTPA; D)c | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Acquired pulmonary alveolar proteinosis/autoimmunec | 2 | 0 | 1 | 1 | 0 | 0 | 0 |
| Histology with surfactant dysfunction disorder without a recognised genetic aetiology | |||||||
| Chronic pneumonitis of infancy | 10 | 6 | 3 | 0 | 1 | 0 | 0 |
| Desquamative interstitial pneumonitis | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Nonspecific interstitial pneumonia | 4 | 0 | 3 | 1 | 0 | 0 | 0 |
| Pulmonary fibrosisc | 1 | 0 | 0 | 0 | 0 | 0 | 1 |
| Congenital multisystemic disorders and other genetic diseases | |||||||
| STING-associated vasculopathy with onset in infancy (TMEM173-STING; D)c | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| ILD associated to dyskeratosis congenita (DKC1; D)c | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| Filamin A mutations (FLNA; D)c | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| Multisystemic smooth musle dysfunction (ACTA2; D)c | 1 | 1 | 0 | 0 | 0 | 0 | 0 |
| FARSB, MARS and other ARS mutationsc (a) (R and D) | 1 | 0 | 0 | 1 | 0 | 0 | 0 |
| Pulmonary alveolar proteinosis due to GMCSF receptor deficiency (CSF2RA, CSF2RB; D)c | 1 | 1 | 0 | 0 | 0 | 0 | 0 |
| COPA syndrome (COPA; D)c | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| ILD associated to Arthrogryposis-renal dysfunction-cholestasis syndrome (VPS33B; D)c (b) | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| Alveolar haemorrhage syndromes | |||||||
| Acute idiopathic pulmonary haemorrhage of infancyc | 4 | 0 | 4 | 0 | 0 | 0 | 0 |
| Disorders not specific to infancy | |||||||
| Disorders of the normal host | |||||||
| Hypersensitivity pneumonitis | 16 | 0 | 1 | 1 | 1 | 9 | 4 |
| Chronic eosinophilic pneumonia | 4 | 0 | 0 | 1 | 1 | 1 | 1 |
| Disorders related to systemic disease processes | |||||||
| Rheumatoid arthritis | 3 | 0 | 0 | 1 | 2 | 0 | 0 |
| Mixed connective tissue disease | 5 | 0 | 0 | 0 | 1 | 3 | 1 |
| Scleroderma | 11 | 0 | 0 | 1 | 1 | 1 | 8 |
| Granulomatosis with polyangiitis | 2 | 0 | 0 | 0 | 0 | 0 | 2 |
| Systemic lupus erythematosus | 4 | 0 | 1 | 0 | 0 | 0 | 3 |
| Antiphospholipid antibody syndrome | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| Goodpasture Syndrome | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| ILD in inflammatory bowel disease (Crohn) | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| ILD in dermatomyositis | 5 | 0 | 0 | 0 | 2 | 1 | 2 |
| Behcet disease | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| Churg–Strauss syndrome | 2 | 0 | 0 | 0 | 0 | 2 | 0 |
| Chronic alveolar haemorrhage, microscopic polyangiitisc | 1 | 0 | 0 | 0 | 1 | 0 | 0 |
| ILD associated to leukocytoclastic vasculitisc | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| Pulmonary lymphomatoid granulomatosisc | 2 | 0 | 1 | 0 | 0 | 1 | 0 |
| Sarcoidosis | 4 | 0 | 0 | 0 | 3 | 0 | 1 |
| Langerhans cell histiocytosis | 9 | 0 | 4 | 1 | 1 | 1 | 2 |
| Tuberous sclerosisc | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| Storage diseases | |||||||
| Niemann–Pick disease (c) | 8 | 0 | 1 | 0 | 7 | 0 | 0 |
| Hermansky–Pudlak syndrome | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Gaucher disease | 2 | 0 | 2 | 0 | 0 | 0 | 0 |
| ILD associated to Mucolipidosis type IIc | 1 | 0 | 0 | 1 | 0 | 0 | 0 |
| Hurler syndrome (MPS I)c | 1 | 0 | 0 | 0 | 1 | 0 | 0 |
| Hunter syndrome (MPS II)c | 1 | 0 | 0 | 0 | 1 | 0 | 0 |
| Other alveolar haemorrhage syndromes | |||||||
| Idiopathic pulmonary haemosiderosisc | 24 | 1 | 3 | 4 | 9 | 5 | 2 |
| Disorders of the immunocompromised host | |||||||
| ILD associated to Chronic granulomatous diseasec | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| Granulomatous Lymphocytic Interstitial Lung Diseasec | 3 | 0 | 0 | 0 | 0 | 1 | 2 |
| ILD associated to Severe combined immunodeficiency (SCID) due to ADA deficiencyc | 1 | 0 | 1 | 0 | 0 | 0 | 0 |
| ILD associated to Medullary aplasiac | 1 | 0 | 0 | 0 | 0 | 0 | 1 |
| ILD in LRBA deficiencyc | 2 | 0 | 0 | 0 | 1 | 1 | 0 |
| ILD associated to Autoimmune lymphoproliferative syndrome (ALPS)c | 1 | 0 | 0 | 0 | 1 | 0 | 0 |
| ILD related to Gain-of-function mutation in STAT1c | 2 | 0 | 0 | 0 | 2 | 0 | 0 |
| Disorders related to therapeutic intervention: drugs | 9 | 0 | 0 | 0 | 1 | 2 | 6 |
| Disorders related to therapeutic intervention: radiotherapy | 3 | 0 | 0 | 0 | 0 | 2 | 1 |
| Lymphatic disorders (Disorders masquerading as ILD) | |||||||
| Primary lymphangiectasia | 7 | 1 | 4 | 2 | 0 | 0 | 0 |
| Diffuse pulmonary lymphangiomatosis | 2 | 1 | 0 | 1 | 0 | 0 | 0 |
| Other entities | |||||||
| Lymphocytic interstitial pneumoniac | 8 | 1 | 1 | 0 | 3 | 2 | 1 |
| Follicular bronchiolitisc | 2 | 0 | 0 | 0 | 1 | 1 | 0 |
| Bronchiolitis obliterans Organising pneumoniac (d) | 16 | 0 | 3 | 0 | 3 | 5 | 5 |
| Alveolar microlithiasisc | 6 | 1 | 1 | 0 | 2 | 1 | 1 |
| Pleuroparenchymal fibroelastosisc | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| Lung disease with unknown cause | 37 | 13 | 20 | 1 | 2 | 0 | 1 |
| Total number of cases | 381 | 65 | 144 | 22 | 52 | 51 | 47 |
Data from 2019. Adapted from Deutsch GH, et al. Pathology Cooperative Group; ChILD Research Co-operative. Diffuse lung disease in young children: application of a novel classification scheme. Am J Respir Crit Care Med. 2007;176:1122.
Added entity not included in the original classification. (a) The only case in this group corresponds to a FARSB mutation. (b) Characterised by arthrogryposis, renal tubular dysfunction and neonatal cholestasis, and platelet abnormalities and may be associated to pulmonary haemorrhage (personal observation). (c) Niemann–Pick type B (SMPD1) 5, Niemann–Pick type A/B (SMPD1) 1, Niemann–Pick type C1 (NPC1) 2. (d) Only cases of BOOP non-related to transplantation or infection are included, other types of obliterans bronchiolitis are not included. Excluded entities from the original classification: pulmonary hypoplasia, prematurity-related chronic lung disease, infectious and postinfectious processes, aspiration syndromes, malignant infiltrates, opportunistic infections, disorders related to transplantation and rejection syndromes, veno-oclusive disease, congestive changes related to cardiac dysfunction. R: recessive biallelic mutation; D: dominant mutation; STFPB: surfactant protein B; STFPC: surfactant protein C; ABCA3: ATP-binding cassette 3; TTF1: thyroid transcription factor 1; SLC7A7: Solute Carrier Family 7 Member 7; STFPA: surfactant protein A; STING: stimulator of interferon; DKC1: Dyskerin Pseudouridine Synthase 1; FLNA: Filamin A; ACTA 2: Actin alpha 2; FARSB: Phenylalanine-tRNA synthetase Betachain; MARS: methionyl transfer RNA synthetase; GMCSF: Granulocyte-macrophage colony-stimulating factor; CSF2RA: Colony Stimulating Factor 2 Receptor Subunit Alpha; CSF2RB: Colony Stimulating Factor 2 Receptor Subunit Beta; COPA: non-clathrin-coated vesicular coat protein A; VPS33B: Vacuolar protein sorting 33. ILD: Interstitial Lung Disease; MPS: Mucopolysarcharidosis; SCID: severe combined immunodeficiency; ADA: adenosine deaminase; STAT1: Signal transducer and activator of transcription 1; m: months; y: years.
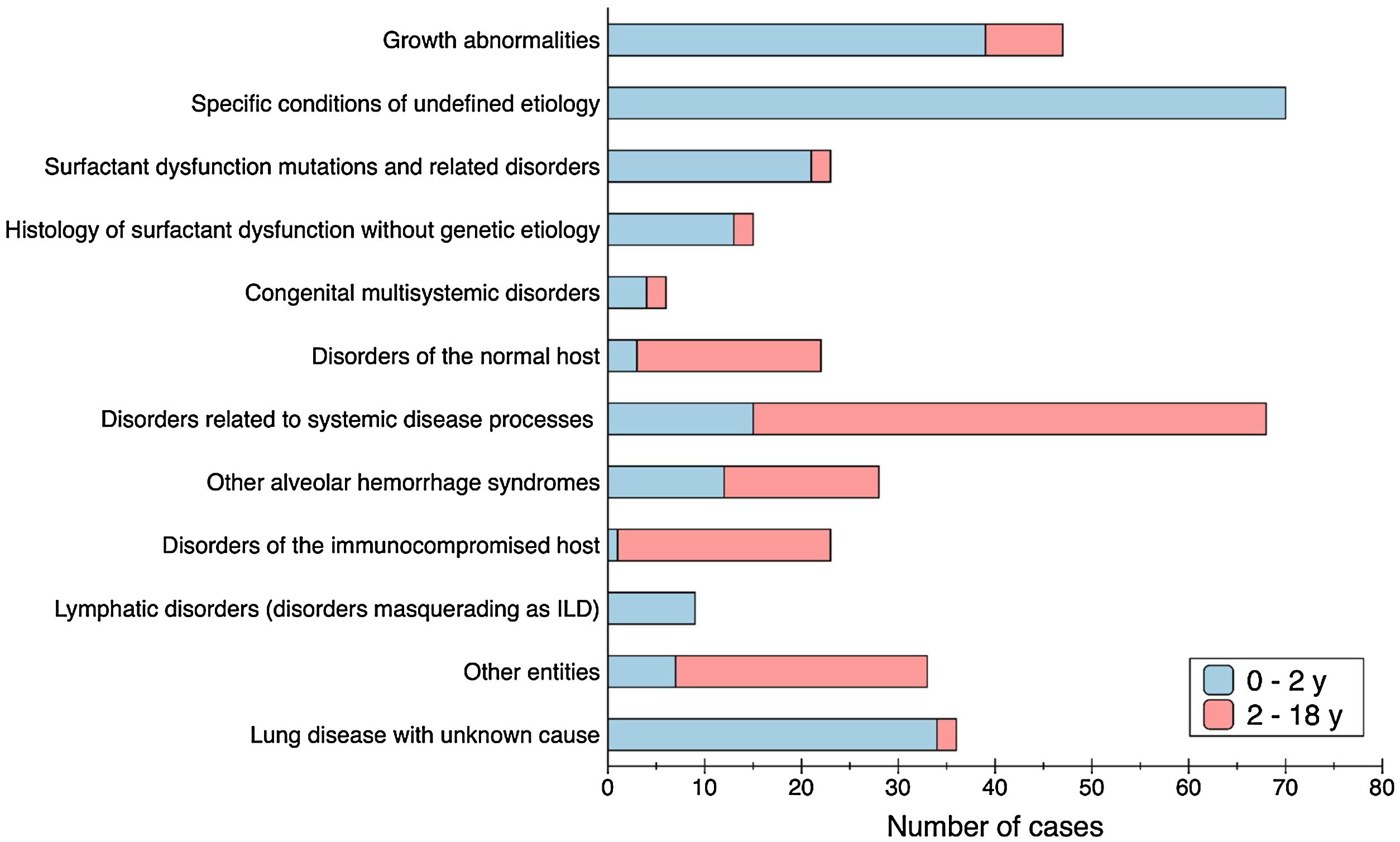
There were different types of disorders seen in children 2–18 years of age compared with children 0 to 2 years of age. Fig. 1 shows the asymmetric distribution of disease categories according to age at presentation. Age distribution at the onset of the disease was not homogeneous as well, and younger patients were more affected: children under 1 year old were the most common age group representing the 54.8% (209/381), among them a third were neonates (65/209); cases under 2 years of age predominate compared with older children (231 vs 150 cases); 74% of patients started their first symptoms before 5 years of age. Most frequent cases stratified by age group were: primary pulmonary interstitial glycogenosis (17/65) and lung disease of unknown cause (13/65) in neonates, neuroendocrine cell hyperplasia of infancy (44/144) in infants from 1 to 12 months old; idiopathic pulmonary haemosiderosis in children from 1 to 5 years old (13/74), hypersensitivity pneumonitis in children from 5 to 10 years old (9/51); and scleroderma in older than 10 years old (8/47).
The 9.7% of cases (37/381) had a disease-causing mutation (Table 4), and the same number of cases (37/381) had no clear aetiology and were reported as lung disease with unknown cause (Table 3).
Disorders With an Identified Gene Mutation Which Lead to the Diagnosis of chILD.
| Disorders | n |
|---|---|
| Surfactant dysfunction mutations and related disorders | 20 |
| Surfactant protein B mutations (SFTPB) | 2 |
| Surfactant protein C mutations (SFTPC) | 9 |
| ABCA3 mutations (ABCA3) | 5 |
| Brain-lung-thyroid syndrome (NKX2-1/TTF-1) | 1 |
| Lysinuric protein intolerance (SLC7A7) | 3 |
| Congenital multisystemic disorders | 7 |
| STING-associated vasculopathy with onset in infancy (TMEM173-STING) | 1 |
| ILD associated to dyskeratosis congenita (DKC1) | 1 |
| Filamin A mutations (FLNA) | 1 |
| Multisystemic Smooth Musle Dysfunction (ACTA2) | 1 |
| FARSB, MARS and other ARS mutations | 1 |
| Pulmonary alveolar proteinosis due to GMCSF receptor deficiency (CSF2RA, CSF2RB) | 1 |
| ILD associated to Arthrogryposis-renal dysfunction-cholestasis syndrome (VPS33B) | 1 |
| Disorders related to systemic disease processes | 10 |
| Storage disease: Niemann–Pick disease (SMPD1, NPC1) | 8 |
| Storage disease: Gaucher disease | 2 |
| Total number of cases | 37 |
Data from 2019. STFPB: surfactant protein B; STFPC: surfactant protein C; ABCA3: ATP-binding cassette 3; TTF1: thyroid transcription factor 1; SLC7A7: Solute Carrier Family 7 Member 7; STING: stimulator of interferon; DKC1: Dyskerin Pseudouridine Synthase 1; FLNA: Filamin A; ACTA 2: Actin alpha 2; FARSB: Phenylalanine-tRNA synthetase Betachain; MARS: methionyl transfer RNA synthetase; GMCSF: Granulocyte-macrophage colony-stimulating factor; CSF2RA: Colony Stimulating Factor 2 Receptor Subunit Alpha; CSF2RB: Colony Stimulating Factor 2 Receptor Subunit Beta; VPS33B: Vacuolar protein sorting 33.
During the study period there were two deaths, 2 neonates. One with pulmonary interstitial glycogenosis associated to pulmonary hypertension and the other with chronic neonatal lung disease and pulmonary hypertension, who had a SMAD9 mutation. One patient with STING-associated vasculopathy with onset in infancy (SAVI) received a lung transplant during this period.
DiscussionThis is the first study to describe incidence and prevalence of chILD in a Mediterranean population, providing an average incidence rate of 8.18 new cases/million of children per year, and an average prevalence rate of 46.53 cases/million of children during the study period 2018–2019.
Despite these diseases are still under-diagnosed, due to nonspecific clinical manifestations and their low frequency, we have been able to collect a relevant number of cases, which emphasises the progress made in identifying and categorising them over the last years.
We found a higher number of new and prevalent cases compared with series published some years before in other locations. Our incidence was between 5 and 6 times higher than the previously found in Germany11 and Australia,13 and 13 times higher than the prevalence found in the UK and Ireland.10 As it has been stated before, these reported frequencies might be underestimated.1,5,27 Even though some of the reasons rely on misdiagnosis, lack of common registers at the time these data were analysed, and outdated diagnostic criteria, as some neonatal disorders were not considered, and new entities have been identified since then.
We think that our finding of much higher incidence and prevalence might be due to greater notice and understanding of these conditions over the last years, and because there has been an increased clinician awareness as we have been in contact with all the clinicians monthly to prospectively collect cases. Lastly, European clinical research projects in this specialised area, such as child-EU (European Management Platform for Childhood Interstitial Lung Disease), COST (European Cooperation in Science and Technology), and CRC-ERS (European Respiratory Society Clinical Research Collaboration), might have contributed. Nevertheless, ethnographic differences cannot be ruled out.
The main characteristics of diffuse pulmonary diseases in children are their great heterogeneity among all the entities they encompass and their low prevalence. This heterogeneity and uncommon frequency were highly represented in our cohort, with 65 different disorders reported, and 24 entities with only one patient listed in each of them.
Regarding categorisation, we adapted the classification first published by Deutsch et al. in 2007,2 to include more entities, some described afterwards (Supplemental Table 1). In a similar way to what has been described previously, different types of disorders were seen in children 2–18 years of age compared with children 0–2 years of age.4,7,28–30 In infants less than 2 years of age, specific conditions of undefined aetiology and growth abnormalities were the largest categories, while for older children, disorders more common in infancy constituted a small group, and the predominant category was disorders associated to systemic diseases. Along with the distinct type of conditions, the distribution of cases by age at the onset of the disease was not homogeneous, with a predominance of cases at younger ages, as it was also reported before in the previous epidemiological studies mentioned.10,11,13 When analysing frequencies by age group, we found a great difference between the group less than 2 years of age (231 cases) with a prevalence rate of 220.29 cases/million (CI 95% 192.8–250.6), compared to the group 2–18 years old (150 cases) which rate was 22.77 cases/million (CI 95% 19.27–26.71). Children under 1 year of age were the most common group representing more than half of total cases (54.5%).
When facing with rare entities, receiving a proper diagnosis can be one of the greatest challenges. In our cohort, almost 10% of patients could not be categorised, and were reported as lung disease with unknown cause (37/381). Besides, there were 15 cases (4%) classified in the category “Histology with surfactant dysfunction without a recognised genetic aetiology” which were diagnosed only by the pathology pattern. Therefore, it results in a greater number of cases not completely well defined. This high number of undiagnosed or not fully identified cases indicates that it is still necessary to improve chILD identification. Current guidelines propose a stepped algorithm for the diagnosis approach including chest CT scan, lung function tests, genetic tests, bronchoscopy with bronchoalveolar lavage, and lung biopsy.3,31 Given the invasiveness of lung biopsy and despite it is the gold standard for some diagnoses, it is currently suggested that some diseases may not require histopathological confirmation,6,29 giving more relevance to the approach based upon genetic tests. Our study highlights the use of genetic tests as a relevant diagnostic tool, since almost 10% of cases (most of them infants) had disease causing mutations identified, leading to the diagnosis. We believe that the percentage of cases with a genetic diagnosis will increase as new genes are described. In this sense, collaboration between centres specialised in genetic studies can contribute to improve diagnosis confirmation.
One limitation of this study was that the focus was mainly on epidemiological data, and we did not collect information related to diagnostic workup of the whole cohort. We could not analyse the percentage of cases confirmed by lung biopsy or only by clinical symptoms and CT scan abnormalities. Due to the epidemiologic nature of our study, we had no independent confirmation of the diagnosis made in the different centres.
Over the two years period of the study, only two deaths were reported, both were neonates, which yielded a mortality rate of 0.95% in infants (2/209). These figures contrast with previous publications where they found higher mortality in the group of infants: 5 deaths out of 21 infants (24%),11 5 deaths out of 63 cases less than 2 years old (8%).13 Our low mortality rate could be explained by the serendipity absence of some lethal diseases concurring with the study period, but also because maybe these patients died in neonatology services without interaction with paediatric pulmonology units or might not have been diagnosed and therefore not registered.
Another limitation of the study was the need for volunteering collaboration, which could lead to under-reporting of all true cases. There were no additional funds or incentives available for the centres.
The reference population used for the statistical analysis was extracted from census data of the National Statistics Institute of Spain, which represents a quantitative measurement of the resident population in each region of Spain, disaggregated according to age. As the study was conducted with the collaboration of 51 paediatric pulmonology units situated all over the country, the reference population considered was the sum of residents in each region with a participating hospital in the study. For those hospitals that covered the population of its local area and were also centre of reference for other regions, we considered all residents in those regions as the reference population as well. In this way, the reference paediatric population of the study was 7,644,155 in 2018 and 7,636,093 in 2019, the 91.7% of the global population up to 18 years old in Spain for both years, which is a broad population to consider the results of our study significant.
Relevant progress has been accomplished over the last two decades in classification, clinical management and research in the field of chILD, as well as in developing international organisational structures to enhance the care of affected children. In this regard, the results obtained in our multicentre study contribute to this ongoing improvement. Understanding of the specific disorders is likely to evolve progressively and larger international studies with a central database as the chILD-EU registry16 using the same diagnostic criteria are expected to yield more accurate results in the future.
Grant SupportATV was supported by a grant from the Spanish Society of Paediatric Pulmonology and a Short Term Scientific Mission of the Cost CA 16125 ENTeR-chILD. This work was supported by a grant from the Spanish Society of Pneumology and Thoracic Surgery (SEPAR 2017/492). AMG was supported by a grant from the project HCQ4Surfdefect, in E-Rare-3, the ERA-Net for Research on Rare Diseases (Acciones complementarias en Salud, Instituto Carlos III, Madrid, Spain, AC16/00027) and Cost CA 16125 ENTeR-chILD.
Conflict of InterestThe authors declare that they have no conflict of interest.
We want to thank the collaboration of all members of the ChILD-Spain Group and all physicians who participated in the clinical care of these patients. We want to thank Dr. Vicente Plaza for the review within the framework of the SEPAR Mentor program. This work has been carried out under the doctoral program of Paediatrics, Obstetrics and Gynaecology of the Universitat Autònoma de Barcelona. MG, AMG and ATV are partners of ERN-LUNG.
Anselmo Andrés, Verisima Barajas, Monserrat Berrocal Castañeda, Mª Araceli Caballero Rabasco, Maria Chine, Fernando Echávarri Olavarría, María Ofelia Fernández de la Cruz, Gemma Garcia del Cerro, Yolanda González Jiménez, David Gómez Pastrana, Ramón Gutiérrez, Elena Hierro Delgado, Javier Korta, Alejandro López Neira, Orlando Mesa, Marianela Marcos Temprano, Mª del Mar Martínez Colls, Luís Moral, Laura Moreno-Galarraga, David Naranjo Vivas, Mª Jesús Navarro, Andreu Peñas Aguilera, Elena Pérez Belmonte, Santiago Pérez Tarazona, Pilar Robles, Teresa Romero Rubio, Santiago Rueda Esteban, María Teresa Rubí Ruiz, José Sirvent, Amalui Vásquez Pérez, Carlos Zabaleta Camino.
The following are the supplementary data to this article:





www.publicationethics.org.




Archivos de Bronconeumología follows the Recommendations for the Conduct, Reporting, Editing and Publication of Scholarly Work in Medical Journals