


La lesión pulmonar aguda es una enfermedad con alta mortalidad, que afecta a gran cantidad de pacientes y cuyo tratamiento continúa en debate. Recientemente, se ha postulado que la hipercapnia podría atenuar la respuesta inflamatoria durante la lesión pulmonar, lo que le otorgaría un papel específico dentro de las estrategias de protección pulmonar durante la asistencia respiratoria mecánica. En el presente trabajo revisamos la evidencia actual sobre el papel que altos niveles de CO2 en sangre desempeñan en la lesión pulmonar. Concluimos que, si bien existen reportes que demuestran beneficios, evidencia más reciente sugiere que la hipercapnia puede ser nociva, contribuyendo a agravar el daño pulmonar.
Acute lung injury is a disease with high incidence of mortality and its treatment is still controversial. Increasing the levels of CO2 beyond the physiological range has been proposed as a potential protective strategy for patients on mechanical ventilation, as it could moderate the inflammatory response. In this article we review the published evidence on the role of CO2 during acute lung injury. We conclude that although there are reports suggesting benefits from hypercapnia, more recent evidence suggests that hypercapnia could be deleterious, contributing to worsening of the lung injury
Mil quinientos millones de años han pasado desde el origen del universo y desde ese momento hasta hoy, este se expande y enfría1. En sentido contrario, en los últimos años aumenta la evidencia sobre la tendencia al ascenso de la temperatura en nuestro planeta y esto se correlaciona con el aumento en los niveles atmosféricos de CO2, en lo que conocemos como fenómeno de «calentamiento global»2.
Este ascenso, directamente relacionado con la emisión de gases generados por la actividad industrial ya ha sido identificado como generador de efectos nocivos, tanto sobre la vegetación3 como sobre los océanos4, 2 de los componentes fundamentales del ciclo del CO2 en la naturaleza.
Los mamíferos, particularmente el hombre, conviven con la necesidad de generar compuestos de alta energía consumiendo oxígeno y produciendo, como contrapartida, anhídrido carbónico. La producción de CO2 es reflejo de la actividad metabólica celular y, por lo tanto, variable en función del órgano estudiado5. Independientemente de estas diferencias, a nivel plasmático el CO2 es mantenido dentro de un estrecho rango (40±5mmHg). Más aún, su acumulación genera rápidamente modificaciones del equilibrio ácido-base, por lo que existen múltiples mecanismos que mantienen los niveles de CO2 plasmático en ese rango fisiológico. En líneas generales, el transporte de CO2 desde su producción mitocondrial hasta su eliminación en el espacio alveolar pulmonar implica la acción de dos mecanismos: difusión simple y difusión facilitada debido a la acción de la anhidrasa carbónica que permite el transporte en paralelo de bicarbonato6. En este contexto, durante décadas ha prevalecido el concepto de que el CO2 constituye un elemento de desecho que debe ser eliminado en función de los disturbios ácido-base que ocasiona pero que, en sí mismo, no es un elemento perjudicial para el organismo. De esta manera, ha sido clásicamente asociado a patologías respiratorias en las que existe una disminución de la renovación del gas alveolar (debilidad muscular, obstrucción pulmonar crónica, ataque agudo de asma, por citar algunos). Sin embargo, en los últimos años el anhídrido carbónico ha estado vinculado con diversos procesos biológicos diferentes al equilibrio entre ácidos y bases y, por lo tanto, ha surgido una importante cantidad de información novedosa al respecto.
La lesión pulmonar aguda es una entidad grave, que requiere un sistema de atención y monitoreo complejos. Sus mecanismos de producción y estrategias terapéuticas están en constante revisión y recientemente, el CO2 ha sido propuesto como una posible herramienta terapéutica7.
En el presente trabajo revisaremos cual es la evidencia científica sobre los efectos del CO2 durante la lesión pulmonar aguda.
Hipercapnia permisivaEl efecto deletéreo de la sobredistensión alveolar durante la ventilación mecánica se publicó en la década de los 608, aunque no se reconoció como un problema clínico en ese momento. Más adelante, la utilización de imágenes tomográficas pulmonares permitió elaborar el concepto de baby lung9 y demostrar cuán irregular es la distribución de la lesión pulmonar.
A mediados de los 80 es cuando surge el término «volutrauma», el cual jerarquizó el rol del volumen pulmonar máximo (y no la presión pulmonar máxima) como mejor equivalente de estrés mecánico y, por lo tanto, generador de la lesión parenquimatosa10,11. No fue hasta fines de los 90 que se impuso la idea de «biotrauma» como mecanismo global de lesión pulmonar asociado a la ventilación mecánica12 y, por lo tanto, la necesidad de optimizar el patrón ventilatorio como estrategia «protectora» del tejido pulmonar.
Este concepto se ha visto posteriormente consolidado con los resultados aportados por el ARDSnet en relación a la mortalidad en función del volumen corriente utilizado13.
Con esta visión de protección mecánica del tejido pulmonar, la ventilación con volumen corriente cercano a 6ml/kg de peso ideal resultó en una reducción de la ventilación alveolar y la retención de niveles variables de CO2 en plasma. El debate posterior a este ensayo multicéntrico se ha enfocado en desvelar los mecanismos involucrados en la reducción de la mortalidad más allá de la reducción del volumen corriente. Existen muchas áreas de incertidumbre a propósito del papel que juega la respuesta inflamatoria sobre la micromecánica pulmonar, la capacidad de reparación celular y la preservación de la interacción epitelio-endotelio alveolar en este contexto. La hipercapnia y la acidosis que ella determina han sido dos de los candidatos más intensamente evaluados como responsables, tanto del daño como de la protección, y los resultados, hasta ahora, no son definitivos.
Inflamación y CO2La inflamación genera marcados cambios a nivel celular y muchas veces las acciones terapéuticas determinan respuestas contradictorias e inesperadas.
Uno de los mecanismos más extensamente estudiados ha sido la generación de especies reactivas de oxígeno debido a su participación en la respuesta frente a la agresión, reparación y muerte celular14.
El anhídrido carbónico reacciona tanto con las especies reactivas de oxígeno como con las especies derivadas de nitrógeno. En un medio acuoso esta reacción se presenta como protectora y reductora del daño oxidativo. Sin embargo, en un entorno no polar de membranas biológicas, el CO2 forma parte de los mecanismos que conducen a nitración proteica y daño oxidativo15. Este rol dual podría explicar la aparente contradicción entre publicaciones que atribuyen al CO2 tanto un papel protector16 como lesivo17 en modelos experimentales de daño pulmonar.
Los mediadores de la respuesta inflamatoria, particularmente las citoquinas, han sido objeto de múltiples estudios, tanto sobre el origen18,19 como del tratamiento20 de los estados de inflamación sistémica.
Existe un acuerdo bastante generalizado en que inicialmente son las citoquinas liberadas por el epitelio alveolar las que comandan la activación y reclutamiento de neutrófilos hacia el espacio alveolar, alterando la permeabilidad endotelial, amplificando la respuesta inflamatoria local e iniciando la etapa de edema pulmonar clínicamente aparente20. Si bien existe considerable evidencia sobre su efecto inmunosupresor (o al menos inmunomodulador)21, el anhídrido carbónico también ha sido reportado como parte de un mecanismo de activación de la respuesta inmune22. Aunque mayoritariamente se acepta su papel inhibidor de la respuesta inmunitaria, esto no resuelve el dilema debido a que mantener un estado de inmuno debilidad no parece la estrategia más razonable para una patología con un importante componente infeccioso23.
En el otro extremo, de confirmarse su rol pro inflamatorio, las consecuencias de la hipercapnia sobre la lesión pulmonar aguda serían un mayor deterioro anatómico y funcional del tejido pulmonar.
Reparación celular y CO2Existen innumerables publicaciones sobre los mecanismos de daño celular24–26, reparación27 y fibrosis tisular28 durante la lesión pulmonar. Sin embargo, el papel del CO2 en estos mecanismos ha sido poco estudiado.
En estudios in vitro se ha observado que niveles elevados de CO2 inhiben el crecimiento celular29. Más aún, se ha especulado con que la acumulación de ciertos aminoácidos intracelulares sería un mecanismo protector frente a la hipercapnia30. Esta inhibición del crecimiento celular ha llevado a explorar, entre otros fenómenos, cual es el efecto del CO2 sobre la capacidad de reparación de la barrera epitelio-endotelial31.
En este sentido, es creciente la evidencia experimental sobre la disminución de la capacidad de reparación endotelial32 y epitelial33 frente a la agresión en presencia de altos niveles de anhídrido carbónico.
La mayoría de los autores consideran que la condición «sine qua non» para que ocurra remodelación y reparación tisular es la apoptosis20. Este mecanismo de muerte celular es inhibido durante la etapa exudativa del distress, permitiendo que la infiltración por neutrófilos se prolongue en el tejido pulmonar24. Al mismo tiempo, existe un estímulo para la apoptosis de las células epiteliales alveolares34,35 configurando la imagen histopatológica del distress descrita hace muchos años caracterizada por persistente infiltrado inflamatorio junto a pérdida de epitelio alveolar36. Por ejemplo, en un modelo experimental de lesión pulmonar por inhalación, la remoción de CO2 acumulado por la aplicación de un patrón ventilatorio protector redujo las modificaciones histológicas y disminuyó los niveles de marcadores de apoptosis celular37. Esta evidencia, si bien es experimental y limitada, reúne en un solo modelo animal parte de la información aportada previamente sobre el rol del anhídrido carbónico en los procesos de lesión y reparación que se desarrollan a lo largo del distress respiratorio. Más aún, abre una interesante interrogante sobre la utilidad de los sistemas de remoción extracorpórea de CO2 asociados a una estrategia de protección pulmonar en pacientes con distress como dirección a futuro.
Edema pulmonar y CO2El edema pulmonar es uno de los elementos más característicos de la lesión pulmonar aguda y el distress respiratorio38. En el origen del mismo se destaca el aumento de la permeabilidad capilar, aunque existe creciente evidencia sobre el papel preponderante de la disfunción del epitelio alveolar. Mientras que la disfunción del endotelio capilar es responsable del escape de líquido desde el espacio vascular, el epitelio disfuncional es responsable de la disminución de la reabsorción de líquido alveolar y la alteración del surfactante pulmonar.
Existen datos experimentales que apoyan los efectos beneficiosos del CO2 sobre la vasculatura pulmonar y sistémica39. Sin embargo, también se ha observado una alteración de la relación ventilación-perfusión40,41 que, sumado al deterioro de la capacidad de reparación endotelial32, ponen en duda que el CO2 determine un mejor intercambio gaseoso durante la lesión pulmonar.
La reabsorción del edema pulmonar es uno de los principales mecanismos que permiten mantener el espacio alveolar «seco». La misma es la consecuencia del transporte activo de sodio a través de la barrera epitelial gracias a la acción de la Na,K-ATPasa. Ha sido bien documentado como la disminución en la capacidad de reabsorción del fluido alveolar determina la mortalidad de pacientes con edema pulmonar42,43. Muchos de los elementos presentes durante la lesión y distress pulmonar deterioran la reabsorción del fluido alveolar como por ejemplo hipoxia44,45, activación endotelial46 y el estrés mecánico47. Asimismo, la hipercapnia, independientemente de la acidosis, deteriora la reabsorción del fluido alveolar estimulando la endocitosis de la Na, K-ATPasa48,49.
Desde el punto de vista mecánico, la presencia de surfactante pulmonar es clave para disminuir la tensión superficial, aumentar la superficie de intercambio alveolar y reducir la tendencia al colapso del tejido pulmonar al final de la espiración.
Su síntesis, secreción y ensamblado en el espacio alveolar son procesos complejos, que requieren consumo de energía y la puesta en marcha de diferentes vías de señalización intracelular. En diferentes estudios, tanto básicos como clínicos, la pérdida de su capacidad tensoactiva se ha vinculado a la lesión pulmonar y al distress respiratorio. Sin embargo, su reposición con agentes semisintéticos no ha demostrado ser eficaz en la patología respiratoria de pacientes adultos.
En relación a la hipercapnia, la presencia de niveles altos de CO2 en modelos experimentales disminuye la secreción de surfactante sin disminuir el metabolismo celular50. De esta manera se sugiere que la disminución de su producción no obedece a un mecanismo protector de la viabilidad celular por disminución del consumo energético, sino que agrega un nuevo mecanismo de estrés mecánico al tejido pulmonar. Aún más, cuando el tejido epitelial recurre a la conservación y compensación energética en respuesta a la hipoxia, se estimula la síntesis de anhídrasa carbónica IX, que aumenta la hidratación del CO2 y estimula la producción de bicarbonato, como mecanismo regulador del pH intracelular51.
Si bien la hipercapnia ha demostrado tener aspectos prometedores como inmunomodulador y con capacidad de atenuar algunos marcadores de daño pulmonar, la combinación de lesión mecánica, hipoxia y activación inflamatoria como antesala de la hipercapnia como estrategia terapéutica ha llevado a que algunos autores, promotores de su uso, planteen que es necesario explorar aun más antes de iniciar ensayos clínicos7.
Estudios clínicos y CO2Al momento actual, no tenemos estudios en humanos en los que se evalúe el efecto de altos niveles de CO2 durante la lesión pulmonar aguda con protocolos específicamente diseñados a este fin.
La evidencia actual proviene de modelos animales en los que se intenta reproducir el escenario clínico, con resultados contradictorios, como hemos mencionado anteriormente. De todas maneras, los promotores de su uso como herramienta terapéutica se apoyan en evidencia indirecta aportada por el ARDSnet52. En este estudio los autores realizan un detallado análisis estadístico sobre factores asociados a mortalidad de los grupos originalmente distribuidos según el volumen corriente utilizado. Finalmente concluyen que los pacientes hipercápnicos ventilados con volumen corriente de 12ml/kg presentaron menor mortalidad que aquellos con niveles normales de CO2 e igual patrón ventilatorio. Si bien esto es estadísticamente correcto, en el grupo de pacientes ventilados con 6ml/kg no se observaron diferencias de mortalidad en función de su CO2 plasmático. De esta manera resulta difícil separar, en este diseño experimental no elaborado para este fin, si los niveles altos de CO2 pudieran beneficiar a pacientes con distrés respiratorio más allá de la protección aportada por el volumen corriente bajo.
Por otro lado, la hipercapnia ha sido reportada como un factor perjudicial en pacientes con obstrucción pulmonar crónica53, lesión pulmonar41 y recién nacidos54.
Direcciones a futuroLa interacción del CO2 con minerales, vegetales y mamíferos ha inducido cambios profundos en la naturaleza55. De hecho, aceptando que estructuras tan diversas pueden ser modificadas en este proceso por el CO2, muchos investigadores han explorado la posibilidad de que exista algún mecanismo «universal»que reconozca las variaciones en los niveles de anhídrido carbónico. Esa estructura o «sensor» de CO2 no ha sido identificada aún pero son muy interesantes los datos aportados por la investigación básica. Específicamente, se han utilizado modelos experimentales con Caenorhabditis elegans56 y Drosophila57 que han identificado modificaciones inmunitarias, de desarrollo y supervivencia en estas especies, lo que sugiere la existencia de mecanismos de alto impacto biológico influenciados por el CO258. Desafortunadamente, todavía no han sido definidos los mecanismos que regulen estas modificaciones.
En los últimos años se ha reconocido que el anhídrido carbónico es mucho más que un producto de desecho del metabolismo celular. De hecho, se han identificado múltiples efectos de su interacción con estructuras y vías intracelulares, alguna de las cuales son francamente lesivas para el tejido pulmonar (fig. 1).
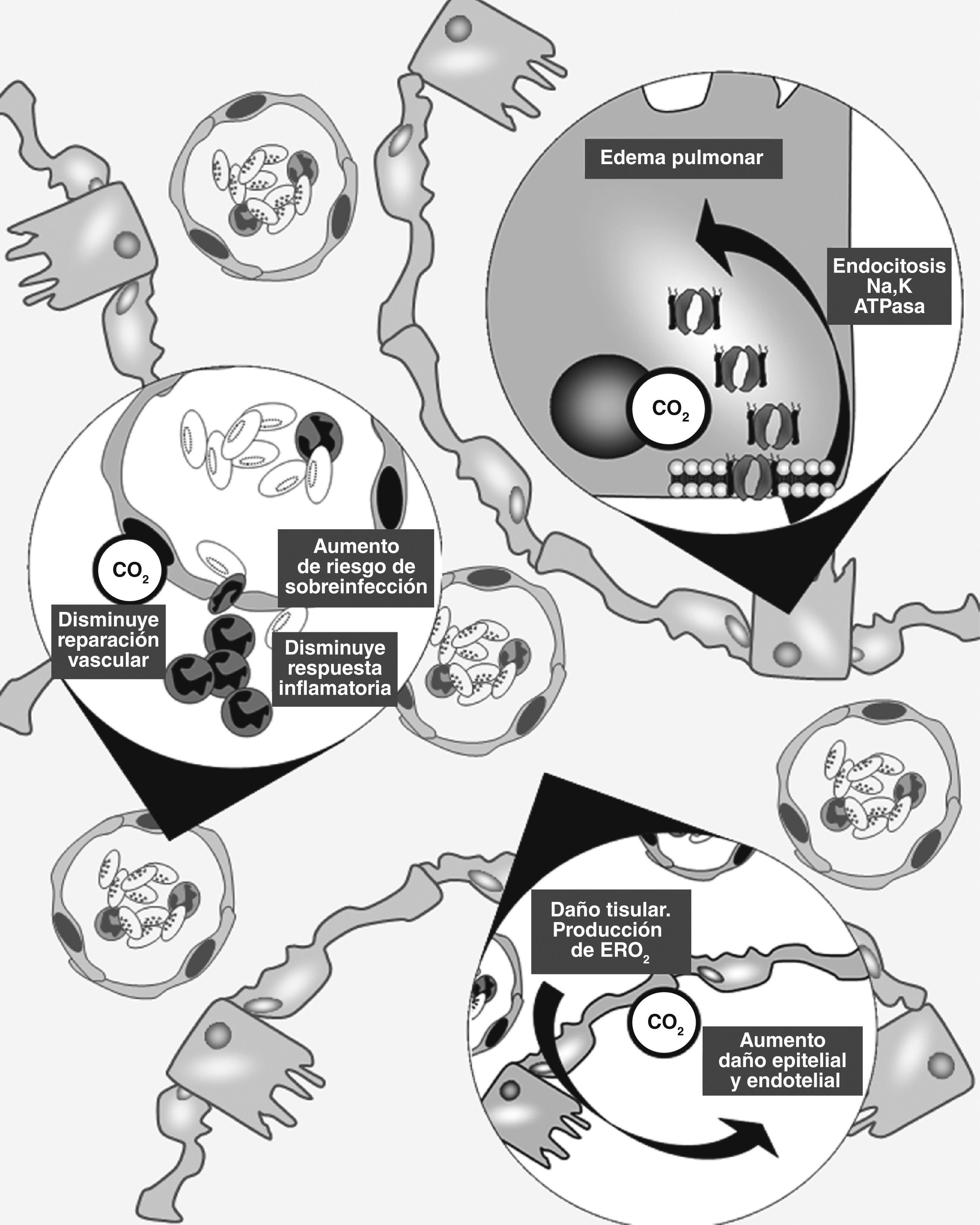
Esquema de los principales mecanismos de lesión pulmonar en los que el CO2 ha sido vinculado. 1) Endocitosis de la Na,K-ATPasa con disminución de la reabsorción de fluido alveolar. 2) Disminución de la respuesta inflamatoria con menor capacidad de reparación tisular y aumento del riesgo a sobre infecciones. 3) Aumento del daño tisular mediado por especies reactivas de O2.
En este camino se han recorrido diferentes etapas: desde la hipercapnia permisiva a la terapéutica y desde la protección hasta la lesión mediada por CO2. ¿Será esta la etapa de la hipercapnia no permisiva?
Sin lugar a dudas que el papel del anhídrido carbónico en el escenario de la lesión pulmonar aguda no está todavía definido. Sin embargo, en función de su importancia a nivel de la biosfera y su capacidad de modular la señalización intracelular, la investigación en esta área está asegurada para el futuro.
Conflicto de interesesLos autores declaran no tener nigún conflicto de intereses.







