



Los alelos deficitarios más frecuentes son los Pi*S y Pi*Z, pero existen también otras variantes deficientes.
En la presente nota clínica se describen los 2 primeros casos detectados en España de déficit de alfa-1-antitripsina (DAAT), resultante de la combinación de un alelo nulo Mattawa con un normal PI*M y con un raro Mmalton.
Ambos casos fueron inicialmente diagnosticados como Pi*MM por isoelectroenfoque (IEE), pero los valores séricos bajos de AAT hicieron sospechar la existencia de alelos deficientes infrecuentes indetectables por IEE, por lo que se realizó un análisis molecular del gen que proporcionó el diagnóstico correcto.
Las incoherencias entre los valores séricos de AAT y el fenotipo deben hacer sospechar la existencia de uno de estos alelos infrecuentes.
The most common deficiency alleles for alpha-1-antitrypsin deficiency (AATD) are Pi*S and Pi*S, but there are also other deficiency variants.
This case report describes the first two cases of AATD detected in Spain resulting from the combination of a null Mattawa allele with a normal PI*M, and a rare Mmalton.
Both cases were initially diagnosed as Pi*MM by isoelectric focusing (IEF), but the low serum AAT values led us to suspect the existence of rare deficiency alleles that were undetectable using this technique, and to performing molecular analysis of the gene, which provided the correct diagnosis.
Inconsistencies between serum AAT values and the phenotype should make one suspect the existence of one of these rare alleles.
Los alelos normales de alfa-1-antitripsina (AAT), presentes en el 85-90% de los individuos, se denominan M, y los deficientes más frecuentes, S y Z (frecuencias respectivas: 10 y 1,7% de la población española)1. Los alelos M, S y Z expresan respectivamente alrededor del 100, del 40 y del 15% de AAT sérica2.
El déficit grave de AAT, definido por niveles séricos por debajo del 35% del valor medio esperado, es una condición rara, generalmente asociada a homocigotos PI*ZZ y con mucha menor frecuencia a combinaciones de alelos Z, S, raros y nulos. Sin embargo, en los últimos años el laboratorio del Registro Español de Pacientes con Déficit de AAT (REDAAT) ha detectado un 1,6% de alelos raros y nulos3 (tasa comparable a la encontrada en Italia, Suiza, Alemania y Estados Unidos)4,5, la mayoría Mmalton, pero también Mheerlen, NullClayton, NullBellingham, Mvhebron, Ybarcelona6-8, etc.
Observación clínicaCaso 1Mujer de 47años no fumadora, con hipertensión arterial esencial, bronquitis de repetición desde los 25años y disnea de esfuerzo en los 3 últimos años.
Su espirometría fue normal (FVC 96%, FEV1 105%, FEV1/FVC 84), pero su capacidad de difusión, DLCO y KCO, fueron del 68 y del 61% de sus valores teóricos respectivos. La TAC torácica presentó acentuación del patrón bronquial sin signos de bronquiectasias ni de enfisema. La función hepática fue normal. La concentración de AAT en suero fue de 73,7mg/dl (referencia: 103-200), compatible con un déficit parcial de AAT, y el fenotipo etiquetado como Pi MM, no concordante con las concentraciones de AAT, por lo que se realizó una secuenciación del gen de la AAT en sangre periférica que detectó una mutación PI Null Mattawa, caracterizada por la inserción de un nucleótido dentro de la región codificadora del exón5, que desplaza el marco de lectura (frameshift mutation) a la posición 376, lo que genera una señal de stop prematura, que finalmente traduce una proteína truncada, muy inestable, que se degrada dentro del hepatocito y resulta indetectable en suero9 (fig. 1).
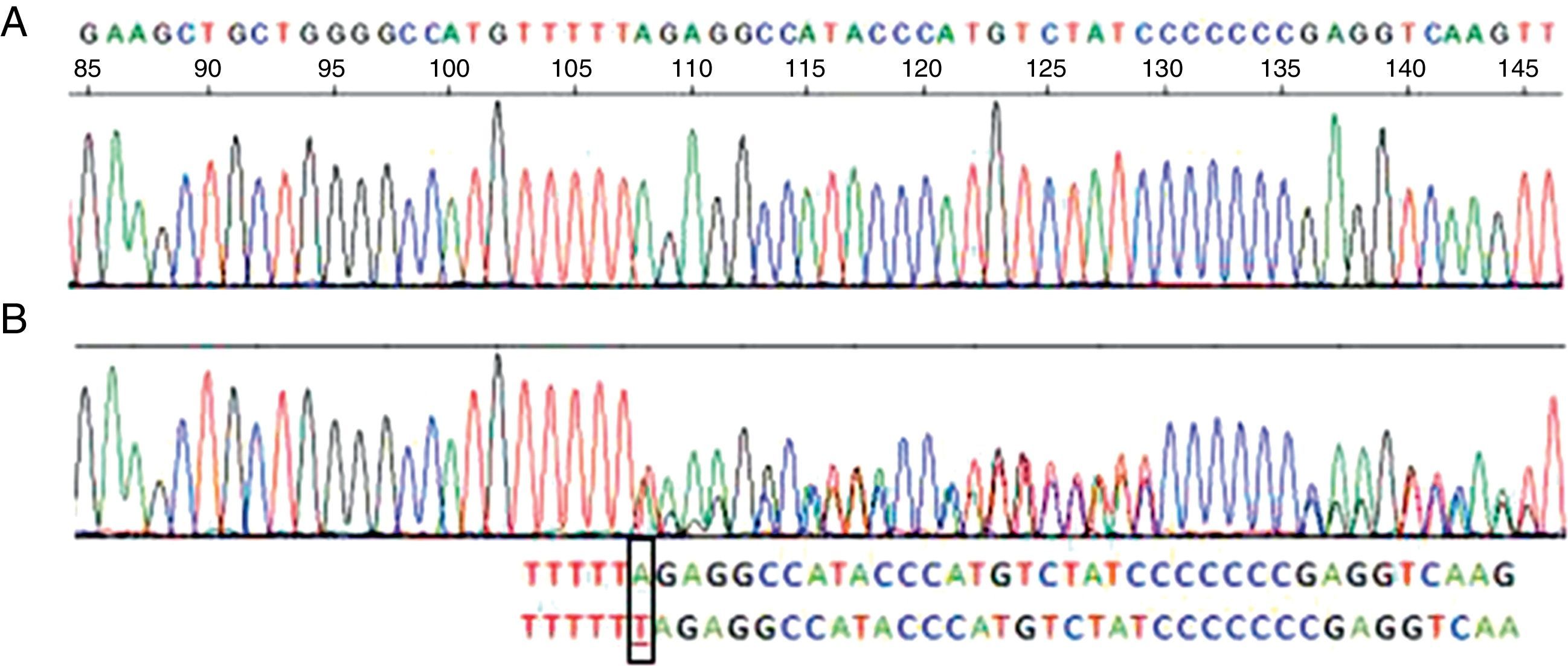
Secuencia correspondiente al exón 5 del gen SERPINA1. A)Secuencia normal. B)Secuencia correspondiente a la paciente, donde se ve la inserción de una timina (T) en vez de una adenina (A) en el codón 376 del exón 5 en heterocigosis, correspondiente al alelo PI-Mattawa. La secuencia codificante del gen SERPINA1 (exones 2 a 5) se analizó utilizando primers previamente descritos para los exones 3 a 5 y primers ex2F 5′ACGTGGTGTCAATCCCTGATCACTG3′ y ex2R 5′TATGGGAACAGCTGG3′ para el exón 2, tomando como referencia comparativa la SERPINA1_Transcript_ENST00000440909.
La paciente tenía además otra variante normal en el exón5, consistente en un cambio de adenina (A) por citosina (C) en el nucleótido 1200 del cDNA, que genera un cambio de aminoácido glutámico (Glu) por aspártico (Asp) en el codón400 (c.1200A>C/p.Glu400Asp), que se correspondía con un alelo M3. Por tanto, se trata de un genotipo heterocigoto PI*M3-Null Mattawa, con niveles séricos de AAT moderadamente reducidos.
Caso 2A partir de la base de datos del REDAAT10 se localizó otra persona portadora del alelo Null Matawa.
Mujer de 67años con antecedentes de tuberculosis pleuropulmonar con curación bacteriológica documentada. Frecuentes episodios de infecciones respiratorias. Parámetros funcionales compatibles con una obstrucción al flujo aéreo grave (FEV1: 620ml [22% del valor teórico], FVC: 1.280ml [39% del valor teórico]). Un TAC de alta resolución mostró enfisema centrolobulillar y paraseptal difuso y bronquiectasias cilíndricas difusas. Nunca se detectaron datos de afectación hepática.
Sus concentraciones de AAT en suero eran de 43mg/dl. El IEE sugirió un fenotipo Pi MM, pero al no corresponderse con la concentración sérica de AAT se realizó un estudio genético, que detectó un alelo PI*Mmalton (deleción del residuo 52)11 y un PI*Null Mattawa, cuya combinación justificó las bajas concentraciones séricas de AAT de la paciente.
DiscusiónLa excepcionalidad de los alelos nulos (prevalencia estimada 100-200 veces inferior que los Z) impide el conocimiento preciso de su impacto clínico y de su verdadera prevalencia. Sin embargo, es importante tenerlos en cuenta, ya que pueden generar confusiones diagnósticas si no se realizan estudios genómicos.
El alelo Mmalton (también denominado Mcagliari y Mnichinan) fue descrito por primera vez en 1987 y, al igual que el gen Z, produce una proteína mal plegada, de la que un 80-90% polimeriza en el hepatocito sin ser secretada a sangre, donde expresa niveles inferiores al 15%, y se asocia con alto riesgo de enfisema pulmonar y de hepatopatía11,12,.
El alelo Mattawa (y en general cualquier alelo nulo) se caracteriza por codificar proteínas con importantes cambios conformacionales, que son degradadas a nivel intracelular sin llegar a polimerizar, y expresan concentraciones indetectables de AAT sérica. Esto hace que los homocigotos conlleven un riesgo muy alto de enfisema, pero no de hepatopatía.
En conclusión, en nuestras pacientes la discordancia entre el fenotipo y las concentraciones séricas de AAT hizo sospechar la existencia de alelos infrecuente, y fue finalmente el análisis molecular del gen el que proporcionó el diagnóstico correcto, tal como se recoge en las recomendaciones2.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Los autores agradecen al Dr. Ignacio Blanco su importante contribución a la redacción de este artículo.







