
Up to 95% of cases related with alpha-1 antitrypsin deficiency (AATD) are associated with the PI*ZZ genotype, while the other 5% are associated with PI*SZ and PI*MZ genotypes or other extremely rare combinations of PI*S or PI*Z with other deficiency or null alleles. These rare alleles account for 1.6% of the deleterious variants recorded in the Spanish Register of Patients with AAT Deficiency, and the most common of these is the PI*Mmalton variant.1 The clinical manifestations of this variant are similar to those of the PI*Z phenotype, and diagnosis is typically delayed by its structural similarity to the Pi M2 allele. We report the case of 2 members of the same family group (Table 1) with a diagnosis of AATD associated with homozygous deficiency PI*Mmalton allele.
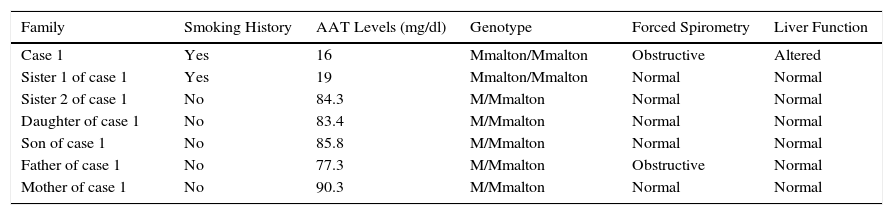
Characteristics of Study Family.
| Family | Smoking History | AAT Levels (mg/dl) | Genotype | Forced Spirometry | Liver Function |
|---|---|---|---|---|---|
| Case 1 | Yes | 16 | Mmalton/Mmalton | Obstructive | Altered |
| Sister 1 of case 1 | Yes | 19 | Mmalton/Mmalton | Normal | Normal |
| Sister 2 of case 1 | No | 84.3 | M/Mmalton | Normal | Normal |
| Daughter of case 1 | No | 83.4 | M/Mmalton | Normal | Normal |
| Son of case 1 | No | 85.8 | M/Mmalton | Normal | Normal |
| Father of case 1 | No | 77.3 | M/Mmalton | Obstructive | Normal |
| Mother of case 1 | No | 90.3 | M/Mmalton | Normal | Normal |
AAT: Alpha-1 antitrypsin.
The index case was a 47-year-old man, native of the island of Gomera (Canary Islands, Spain), with a clinical history of spontaneous pneumothorax in 2005, and former smoker of 30 pack-years, referred to the respiratory medicine clinic due to a 1-year history of dyspnea on moderate exertion (mMRC 2). Lung functional tests showed FEV1/FVC: 0.5; FEV1: 1.73 l (51%); FVC: 3.38 l (77%); DLCO: 73%; and KCO: 75%. High-resolution computed tomography (HRCT) revealed centrilobular and paraseptal emphysema, predominantly in the upper fields. Abdominal ultrasonography found no signs of chronic liver disease, despite mildly elevated transaminases. Complete blood count and IgA, IgM, IgG and IgE levels were within normal limits. Alpha-1 antitrypsin (AAT) was determined by nephelometry, revealing severely reduced levels (16mg/dl), so DNA testing was performed to determine the presence of PI*S and PI*Z alleles, but the result was negative. Given the absence of these deficiency variants and the low AAT levels in serum, we performed a molecular analysis of the SERPINA1 gene, amplifying both coding exonic regions and flanking intronic sequences of exons 4, 5, and 6. This analysis revealed the presence of the homozygous PI*Mmalton (F52del) allele.
The patient's sister, 42 years old, former smoker of 35 pack-years denied any respiratory symptoms. Lung functional tests showed FEV1/FVC: 0.74; FEV1: 2.68 l (96%); FVC: 3.62 l (110%); DLCO: 81%; and KCO: 82%. HRCT, abdominal ultrasonography, and general laboratory tests were all normal. AAT determination revealed severely reduced levels (19mg/dl), and molecular analysis of the SERPINA1 gene detected the homozygous PI*Mmalton allele.
Various AADT patient registries describe the PI*Mmalton allele as the third most common deficiency variant in Spain,2 although this is the first report of its existence in the Canary Islands. Like the Z gene, the Mmalton allele produces improperly folded protein, of which 80%–90% is polymerized in the hepatocyte, expressing levels of less than 15% in blood. The co-existence of emphysema and hepatic cirrhosis is often described in patients with homozygous PI*Mmalton,3,4 although this was absent from our cases; however, the behavior of the heterozygous forms appears to be far more variable. Most PI*M/Mmalton patients have no changes in lung or liver function, in contrast to the PI*Z/Mmalton genotype that appears to be associated with an increased risk of emphysema.5 With regard to liver involvement, Canva et al. described the case of a PI* Mmalton/M patient who developed end-stage liver disease despite no history of hepatitis, alcohol abuse or liver disease in childhood.6 Similarly, Piras et al., found that less than 13% of subjects with either the homozygous or heterozygous PI*Mmalton variant showed evidence of chronic liver disease.7
Our case is remarkable because of the clinical differences observed in our patients, despite their consanguinity and similar exposure to tobacco smoke at a similar intensity, demonstrating different patterns of clinical manifestations of the same PI*Mmalton/Mmalton genotype.
Please cite this article as: Figueira Gonçalves JM, Martínez Bugallo F, Díaz Pérez D, Martín Martínez MD, García-Talavera I. Déficit de alfa-1-antitripsina asociado a la variante Mmalton. Descripción de una familia. Arch Bronconeumol. 2016;52:617–618.







