



La fibrosis quística (FQ) es una enfermedad genética autosómica recesiva que se produce como consecuencia de una alteración del gen cystic fibrosis transmembrane conductance regulator (CFTR)1. Esta alteración determina un transporte anormal de iones, fundamentalmente en las células epiteliales del tracto gastrointestinal y respiratorio2. La detección precoz mediante cribado neonatal a través de la determinación de la tripsina inmunorreactiva ha demostrado beneficios a largo plazo, por ello, generalmente se diagnostica en niños. Sin embargo, estudios recientes indican una prevalencia del diagnóstico de FQ en adultos cercana al 10%3,4. El diagnóstico se establece ante la presencia de criterios clínicos o antecedentes familiares de FQ y la demostración de un funcionalismo anormal del CFTR mediante el resultado de una prueba del sudor o la presencia de 2 mutaciones causantes de la enfermedad4.
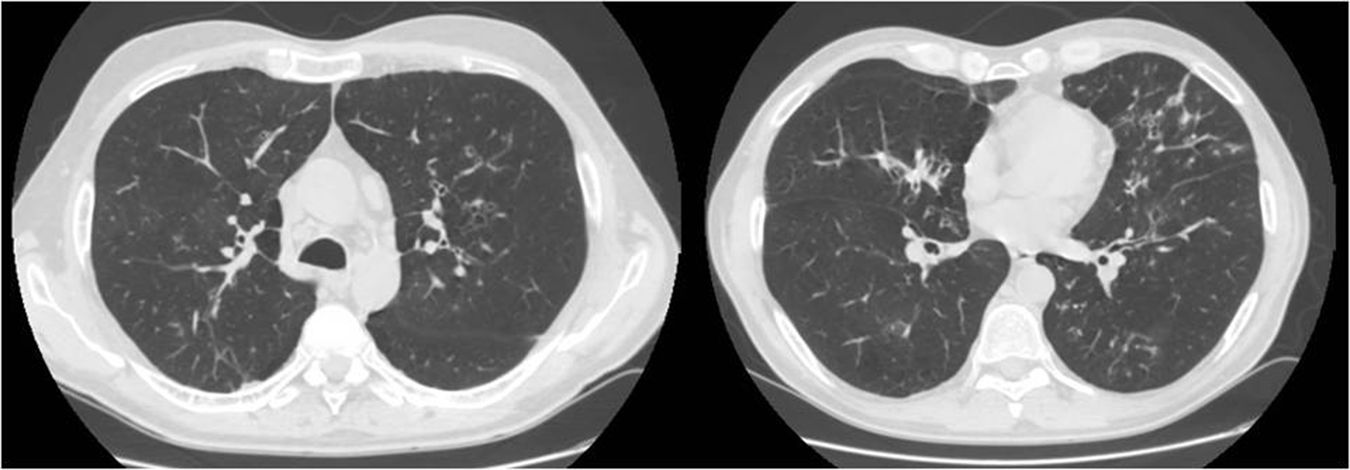
Presentamos el caso de un varón de 52 años de edad, exfumador de 35 paquetes/año, con antecedentes familiares de enfermedad respiratoria, madre afecta de asma y padre afecto de enfisema pulmonar, sin otros antecedentes personales de interés. Ingresa en la unidad de cuidados intensivos por una encefalopatía hipercápnica sin un desencadenante claro, siendo necesaria su intubación y tratamiento con ventilación mecánica invasiva. Previo al ingreso presentaba tos y expectoración habituales y disnea grado 1-2 de la Medical Research Council (MRC) de algunos años de evolución que había empeorado en los últimos meses. Al alta se deriva a la consulta de neumología para completar el estudio y destaca que el paciente mantiene disnea grado 2 de la MRC sin otros datos relevantes. Se solicitan pruebas de función pulmonar, ajustadas a la normativa SEPAR publicada en el año 2002, que revelaron una capacidad vital forzada (FVC) de 2.150cc (51,7%), un volumen espiratorio forzado en el primer segundo (FEV1) de 650cc (19,4%) y una relación FEV1/FVC del 30,5%, una difusión de monóxido de carbono corregido por el volumen alveolar (KCO) del 56% y una prueba de los 6min marcha en la que recorría 576m (98,5% de sus teóricos) aunque desaturando hasta el 76%. Se constató una insuficiencia respiratoria global crónica en la gasometría arterial, cuyos valores eran: pH 7,41, presión parcial de dióxido de carbono (PaCO2) 52mmHg, presión parcial de oxígeno (PaO2) 52mmHg y saturación de oxígeno (SaO2) del 86%, por lo que se mantuvo el tratamiento con ventilación mecánica no invasiva en domicilio. Además se solicitó una tomografía computarizada (TC) de tórax que mostró en el parénquima pulmonar afectación enfisematosa centrolobulillar, áreas bronquiectásicas en campos superiores fundamentalmente y en regiones perihiliares y bases, con formaciones quísticas (bronquiectasias quísticas) y zonas seudonodulares en língula y lóbulos inferiores en relación a impactaciones mucosas (fig. 1). Ante estos hallazgos el paciente fue diagnosticado de enfermedad pulmonar obstructiva crónica (EPOC), insuficiencia respiratoria global crónica y bronquiectasias pendientes de filiar. Con el objetivo de buscar una etiología de las bronquiectasias se solicitaron diversas pruebas complementarias (estudio de inmunodeficiencias, alfa-1-antitripsina, Mantoux…)5 y destacó un test del sudor positivo con un resultado de 84mEq de cloruro por litro que se completó con un estudio genético. El paciente no fue portador de ninguna de las 52 mutaciones que se estudiaron, aunque no se realizó la secuenciación completa del gen. Con todo ello diagnosticamos a nuestro paciente de FQ del adulto.
Las bronquiectasias son la tercera causa de enfermedad respiratoria obstructiva en pacientes adultos. Dado que no se tratan de una enfermedad en sí sino de la consecuencia de otros procesos, es de gran importancia establecer la etiología de las mismas en aras de un mejor abordaje terapéutico5. A pesar de que la prevalencia de bronquiectasias es aproximadamente del 50% en EPOC moderada-grave6, no debemos dar por hecho que son consecuencia directa de esta enfermedad y debemos realizar un diagnóstico diferencial de otros procesos asociados.
En todo el mundo aproximadamente hay 70.000 afectados de FQ7. En las últimas décadas las notables mejoras en el manejo médico, particularmente en lo que se refiere a la afectación pulmonar, ha aumentado la esperanza de vida en estos pacientes8–10 y, actualmente el 40-50% de los pacientes con FQ son adultos3.
En la mayoría de los casos los pacientes presentan la clínica clásica de FQ en la infancia, y el diagnóstico se realiza en el primer año de vida11. Los pacientes que se diagnostican en la edad adulta a menudo tienen una de las llamadas mutaciones «leves» y han conservado alguna actividad del canal del cloro1, son los denominados pacientes con FQ no clásica y suelen tener pocos síntomas y menos órganos/sistemas involucrados.
A pesar de los avances en el tratamiento de la FQ, las manifestaciones respiratorias continúan dominando la clínica característica de los pacientes con FQ, tal y como ocurría con nuestro paciente, y además representan el 95% de la morbimortalidad de esta enfermedad12,13. La afectación pulmonar en los pacientes con FQ clásica incluye bronquiectasias predominantemente en lóbulos superiores, infecciones pulmonares recurrentes, impactación mucoide que se puede asociar a infección crónica bacteriana por Staphylococcus aureus, Pseudomonas aeruginosa o Burkholderia cepacia, aspergilosis broncopulmonar alérgica, neumotórax, atelectasias e incluso hipertensión pulmonar (HTP) y cor pulmonale14. La FQ del adulto se presenta como una FQ no clásica y las manifestaciones pulmonares pueden estar ausentes o tener versiones más leves de los hallazgos típicos.
El paciente adulto ha de recibir una atención integral y personalizada, que debe llevarse a cabo en unidades de FQ con experiencia y multidisciplinarias. El objetivo principal del tratamiento sintomático es retrasar la progresión de la enfermedad. Además, el estudio genético permite suministrar terapias de mutaciones específicas como es el caso del ivacaftor, que modulen el defecto de la proteína CFTR en 9 de las mutaciones conocidas de la FQ y abre camino al desarrollo de otros fármacos modificadores de la enfermedad15.
Este caso muestra como un paciente con enfisema y obstrucción grave al flujo aéreo, que en principio no presentaba dudas diagnósticas, puede sorprendernos arrojando un diagnóstico que, si bien poco frecuente, modifica de forma radical el manejo y el pronóstico, poniendo de manifiesto la importancia del estudio completo de todo paciente con bronquiectasias.







