




Introducción
La enfermedad pulmonar obstructiva crónica (EPOC) está causada fundamentalmente por una reacción inflamatoria frente al humo del tabaco. Sólo un 20-25% de los fumadores desarrolla la enfermedad, pero desconocemos las causas de esta predisposición al desarrollo de la EPOC, si bien es posible que tenga un componente multifactorial que incluya tanto elementos ambientales como de susceptibilidad individual. Como es difícil cuantificar la enfermedad y sus alteraciones en las pequeñas vías aéreas o en el parénquima, donde principalmente se asienta, se sigue midiendo clínicamente por el grado de afectación del volumen espiratorio forzado en el primer segundo (FEV1), que requiere de muchos años de evolución para que se vea afectado e identifica al paciente cuando ya tiene la enfermedad establecida. La EPOC es una enfermedad inflamatoria pero, como su nombre indica, implica la presencia de un componente funcional obstructivo. Sin embargo, el FEV1 puede no ser el mejor método para identificar el inicio de la enfermedad1.
¿Cuándo aparece la EPOC, cuando se identifica el típico infiltrado inflamatorio, cuando existe remodelado o cuando este remodelado produce cambios mecánicos? La EPOC es una enfermedad que tiene una característica afectación del parénquima pulmonar y de las vías aéreas centrales y periféricas, con participación desde su inicio de una alteración asociada en las arterias pulmonares. La afectación característica que presenta el parénquima en esta enfermedad permite definir el enfisema pulmonar, conocido clásicamente por la existencia de un agrandamiento permanente y destructivo de los espacios aéreos distales en ausencia de fibrosis evidente, aunque se admite que existe una remodelación de tejido con incremento neto de colágeno intersticial2. Las vías aéreas también se encuentran siempre afectadas en mayor o menor grado en la EPOC, con hiperplasia de las glándulas mucosas, aumento del número de células caliciformes, zonas de metaplasia escamosa y anomalías ciliares. Sin embargo, son las vías aéreas periféricas las que presentan mayor trascendencia patológica y funcional, con un estrechamiento de su luz como consecuencia de cambios inflamatorios crónicos, impactaciones mucosas, metaplasia de células caliciformes, fibrosis e hipertrofia del músculo liso3.
Desde el humo del cigarrillo a la inflamación
Las causas por las que el humo del cigarrillo lleva al desarrollo de EPOC son múltiples y la patogenia es compleja. Además de la diferencia en la distribución anatómica predominante de la enfermedad en el pulmón, la diferente asociación con la lesión de la vía aérea y las diferencias en la concentración de colágeno y elastina encontradas llevan a pensar en una patogenia diferente para cada variante morfológica que, sin embargo, no ha sido demostrada.
La causa puede ser única pero el proceso es muy prolongado. Un fumador de 20 cigarrillos al día presenta 20 agresiones diarias en el pulmón que reciben una respuesta de los mecanismos de defensa del organismo. Inicialmente responden los mecanismos de defensa de la barrera mucociliar, pero la persistencia de la agresión provoca la rotura de esta barrera y el desarrollo de la respuesta inflamatoria, principalmente de células polimorfonucleares, eosinófilos y macrófagos. Como la agresión persiste, los antígenos depositados en la superficie de las vías aéreas pueden ser transportados al tejido linfático asociado al bronquio por las células dendríticas y desencadenarse la respuesta inmunológica característica de linfocitos T CD8+4.
En el desarrollo y progresión de la EPOC interviene toda una serie de procesos interrelacionados, de entre los que cabe destacar fenómenos de estrés oxidativo, de inflamación y reparación, de acción de las proteasas y de la apoptosis5, todo ello dentro de un sistema en movimiento activado por las fuerzas mecánicas que expanden el pulmón durante el ciclo respiratorio y que podrían colaborar en la destrucción del parénquima pulmonar.
Estrés oxidativo
Cada inhalación de humo de cigarrillo genera 1.015-1.017 radicales libres y, además, el metabolismo oxidativo de otros compuestos en el epitelio bronquiolar y alveolar generaría incluso más radicales libres6. El estrés oxidativo empeoraría la EPOC mediante numerosos mecanismos, entre ellos modificaciones de las moléculas de la matriz extracelular que las harían más sensibles a las proteasas, causarían infiltración neutrofílica, inactivarían los inhibidores de proteasas como la α1-antitripsina7, alterarían la proliferación celular o la reparación alveolar y modularían la respuesta inmunitaria e inflamatoria a través sobre todo de 2 factores de transcripción: el factor de transcripción κB (NF-κB) y la proteína activadora 1, mediadores intracelulares críticos en la cascada inflamatoria. La activación del NF-κB se ha implicado en la patogenia de la EPOC, aunque su papel exacto no está claramente demostrado. Además, la activación del NF-κB está fuertemente asociado con el humo de tabaco, principal factor de riesgo en la EPOC8.
Tras una exposición aguda al humo de cigarro se inicia un mecanismo dependiente de especies reactivas de oxígeno mediante la activación del NF-κB que origina una infiltración de neutrófilos en la vía aérea. De igual modo, se ha observado que el NF-κB está activado en los macrófagos del esputo durante las exacerbaciones de pacientes con EPOC9. Igualmente se ha observado un aumento de la activación de NF-κB en cultivos de tráquea tratados con partículas nocivas10. Además, se ha demostrado que la gravedad de la EPOC está asociada con un incremento de la expresión epitelial del NF-κB11.
Otro factor de transcripción, la proteína activadora 1, es un homo o heterodímero de un grupo de proteínas, Jun y Fos, que, al igual que el NF-κB, puede autorregularse positiva y negativamente a través de los genes que estimulan y aumentan su actividad tras la exposición a tabaco. Además, la administración de antioxidantes antes de la exposición a los agentes nocivos atenúa la activación de proteína activadora 17.
Inflamación en la EPOC
Hogg et al12 fueron los primeros en establecer, en 1968, que la limitación al flujo en la EPOC es debida a un proceso inflamatorio de las vías aéreas periféricas. La inflamación per se podría ser responsable de la limitación al flujo aéreo, bien liberando mediadores de la inflamación que actuarían directamente sobre el músculo liso bronquial, bien mediante la producción de fibrosis peribronquiolar, aunque también podría desempeñar un papel importante la destrucción de las ataduras alveolares que constituyen los septos alveolares conectados con las vías aéreas periféricas y que representan verdaderos tirantes que ayudan a mantener patente la luz de estas vías aéreas13.
En el proceso inflamatorio de la EPOC, las células principalmente implicadas son los neutrófilos, eosinófilos, linfocitos y macrófagos. El contenido y el tipo celular varían en función de que el tipo de muestra obtenida provenga del esputo, del lavado bronquial o broncoalveolar o de biopsia pulmonar, de modo que se encuentran más macrófagos en el lavado, más neutrófilos en el esputo y más linfocitos T en las biopsias14. Los neutrófilos y los macrófagos fueron las primeras células que se implicaron en las anormalidades patológicas del enfisema. Ambos tipos celulares tienen las enzimas proteolíticas necesarias para degradar el parénquima y producir enfisema. En el aumento de células polimorfonucleares y de macrófagos podrían participar modificaciones en el inhibidor α1-proteasa en las vías respiratorias. Las primeras células que responderían a la agresión del tabaco serían los neutrófilos, reclutados por componentes del tabaco como la nicotina, que tiene capacidad quimiotáctica de los neutrófilos humanos15, aunque el mecanismo final por el que se produce este reclutamiento de neutrófilos en el pulmón se desconoce. Es posible que otros componentes del tabaco provoquen una irritación inicial de las células epiteliales que dé lugar a una respuesta inflamatoria temprana con reclutamiento de más neutrófilos y macrófagos. Esto, que se iniciaría en los estadios más tempranos, se puede perpetuar a lo largo de la vida del fumador. Este daño a largo plazo provocaría la aparición de los linfocitos T, que secretarían citocinas que atraerían más macrófagos y neutrófilos, con lo que se generaría un aumento de la proteólisis y del estrés oxidativo.
Varios autores han encontrado una relación del tipo celular y el grado de inflamación existente con el grado de gravedad de la EPOC, lo que indica que la persistencia de la agresión por el tabaco diario va provocando no sólo la continuidad de la respuesta inflamatoria, sino también un incremento de su intensidad. Se ha descrito una clara relación entre el grado de EPOC y el contenido de polimorfonucleares, macrófagos, linfocitos T y células B16, hallazgo consistente con el del aumento de células CD8 y células B a medida que progresa la EPOC encontrado por otros autores17. Esta respuesta inflamatoria parece ser más intensa en el pulmón con predominio de enfisema centroacinar, donde existen más datos de inflamación y de reparación que en el enfisema de predominio panacinar18. Una posible explicación a la existencia de esta continuidad del proceso inflamatorio podría ser el hallazgo del comportamiento anómalo de los macrófagos encontrado en los pacientes con EPOC que muestran una deficiencia en su habilidad de fagocitar las células epiteliales apoptóticas de las vías aéreas19. En el enfisema pulmonar inducido en animales con cloruro de cadmio también se ha observado un aumento del infiltrado inflamatorio con incremento de la presencia de polimorfonucleares y de macrófagos alveolares pocos días después de la inducción de la lesión20. De igual modo, en el modelo de enfisema pulmonar inducido con elastasa también existe un aumento de células en el lavado broncoalveolar que se acompaña de lesión epitelial e infiltración de neutrófilos y macrófagos de la mucosa bronquial21. Estos datos indican que todo proceso destructivo pulmonar asociado a la EPOC se ve acompañado de un proceso inflamatorio de varios tipos celulares, donde los polimorfonucleares y los macrófagos serían los encargados de liberar las proteasas, oxidantes y mediadores como citocinas y quimiocinas, y los linfocitos de mantener activa esta inflamación.
La diversidad de células inflamatorias encontradas en pacientes con EPOC puede ser mejor entendida si se analiza globalmente. Siguiendo la idea de Cosio y Guerassimov14, la reacción inflamatoria más temprana implicaría a los neutrófilos, seguidos de los macrófagos pulmonares en todas las superficies epiteliales del pulmón. Estas células dañarían las células epiteliales y la estructura intersticial subyacente (elastina, colágeno, proteoglucano, etc.). Estas proteínas pueden ser procesadas en péptidos con potencial antigénico que podrían ser reconocidos por linfocitos T, con lo que se iniciarían su activación y proliferación. Las células T activadas pueden a su vez reclutar macrófagos, neutrófilos e incluso eosinófilos hasta el lugar de la inflamación. Visto así, todas las células inflamatorias trabajarían juntas para producir alteraciones de las vías aéreas, la destrucción pulmonar y finalmente la EPOC. Estos datos han permitido elaborar una nueva hipótesis patogénica de la EPOC basada en la existencia de un posible mecanismo autoinmunitario22.
Mediadores de la inflamación
Cuando las células inflamatorias llegan al pulmón, desarrollan allí su capacidad de liberar proteasas, oxidantes y mediadores de la inflamación como las citocinas. Las citocinas son moléculas de señalización que inducen, mediante la interacción con receptores que se encuentran en la superficie celular, movimiento, diferenciación, crecimiento y muerte de muchos tipos celulares. Pueden producir efectos diversos, entre ellos los siguientes: a) iniciación y amplificación de la inflamación; b) activación de las células T independientemente de macrófagos; c) regulación de la maduración y diferenciación de las células dendríticas; d) regulación de la activación y diferenciación de las células T; e) modificación de las estructuras del tejido conectivo, y f) regulación del crecimiento de los vasos sanguíneos. En la tabla I se resume la función de las principales citocinas en las vías aéreas.

En la EPOC se ha demostrado que tienen especial importancia las siguientes citocinas: factor de necrosis tumoral alfa (TNF-α), interleucina (IL) 1β y factor transformador de crecimiento (TGF-β). El TNF-α es una citocina proinflamatoria producida por muchos tipos celulares como macrófagos, células T y células epiteliales. Contribuye a la leucocitosis mediante la liberación de neutrófilos de la médula del hueso e induce la producción de otras citocinas como la IL-8 o RANTES en las células epiteliales respiratorias. También induce la proliferación de fibroblastos que provoca la transcripción de IL-8 a través de NF-κB y aumenta la liberación de IL-8 del epitelio de las vías aéreas y de neutrófilos, además de poder activar a los macrófagos para producir metaloproteasas.
La IL-1β, junto con el TNF-α, participa en la respuesta inflamatoria como quimioatractante de células polimorfonucleares y estimulante de la respuesta de los linfocitos T. La sobreexpresión de IL-1β está además relacionada con el incremento de la deposición de colágeno asociado al proceso de reparación23.
El TGF-β es un factor de crecimiento que influye en la proliferación de fibroblastos, de las células del músculo liso de las vías aéreas y de las proteínas de matriz. Este factor de crecimiento está involucrado en la reparación de la vía aérea y en el proceso de remodelado. Además es quimioatractante de neutrófilos, células T, monocitos y fibroblastos, y regula otros factores como el TNF-α. También modula la citotoxicidad de los macrófagos mediante la supresión de la producción del superóxido y del óxido nítrico24. Además, bloquea la degradación de la matriz reduciendo la síntesis de proteasas e incrementando la síntesis de inhibidores de las proteasas. En pacientes con EPOC se ha observado un aumento de la expresión de TGF-β en el epitelio bronquiolar, además de estar altamente expresado en los macrófagos de las pequeñas vías aéreas25, de modo que se ha encontrado una relación entre el número de macrófagos intraepiteliales y el incremento del TGF-β. Éste también activa la metaloproteasa 9, que activa a su vez el TGF-β, lo cual provoca un enlace entre la fibrosis de las pequeñas vías aéreas y el enfisema en la EPOC. En definitiva, el TGF-β es importante en la transición que hay desde la respuesta inflamatoria inmunitaria hasta el proceso de remodelación tisular26.
Proteasas
La idea del equilibrio proteasa-antiproteasa se basa en varias observaciones clínicas y experimentales, como la presencia de enfisema en el déficit de α1-antitripsina, el hecho de que el suero de pacientes con deficiencia de α1-antitripsina pierda la capacidad para degradar la elastasa, la presencia de una proteasa que podría actuar degradando el pulmón, el aumento de neutrófilos detectado en pacientes con deficiencia de α1-antitripsina y, por último, la observación experimental de inducción de modelos de enfisema por administración de papaína o elastasa. El aumento de proteasas o la disminución de antiproteasas daría lugar a la digestión del esqueleto conectivo del parénquima pulmonar.
Estudios experimentales han mostrado que no solamente la elastasa neutrofílica es la responsable del proceso, sino que también son muy importantes las proteasas secretadas por macrófagos y la falta de síntesis de ciertos proteoglucanos que puede provocar la aparición de enfisema27, así como un importante grupo de metaloproteasas de matriz y sus correspondientes inhibidores tisulares. Sin embargo, muchos fumadores y pacientes con enfermedades pulmonares caracterizadas por un aumento inflamatorio, como la neumonía o el síndrome de distrés respiratorio del adulto, no desarrollan enfisema y, por lo tanto, la hipótesis de la alteración del equilibrio de proteasa/antiproteasa no puede explicar por sí sola el desarrollo y la progresión de la EPOC.
Apoptosis
La muerte celular programada o apoptosis (del griego apo y ptosis, indicativo de la caída de las hojas de un árbol o de los pétalos de una flor) se define como una forma de deceso celular caracterizado por la ejecución de un programa de muerte que tienen todas las células, está codificado genéticamente y es esencial para el desarrollo y mantenimiento de la homeostasis de los tejidos adultos. La apoptosis es un proceso innato y evolutivamente conservado en el cual las células se inactivan, se desensamblan y degradan, de manera coordinada y característica, su propia estructura y componentes, que son fagocitados por macrófagos o incluso por células vecinas para evitar el proceso inflamatorio.
La apoptosis se ha asociado con el enfisema gracias a la instilación de un agente proapoptótico como la caspasa 3, la interrupción de la señal del factor de crecimiento del endotelio vascular (VEGF) o la activación del factor de crecimiento placentario. La inyección intratraqueal de la caspasa 3 en ratones causa la apoptosis de las células de la pared alveolar (principalmente células epiteliales), lo que lleva a la destrucción de la pared alveolar y al aumento de los espacios aéreos28. El VEGF es un factor de supervivencia de las células endoteliales y su retirada da lugar a la apoptosis de éstas. En pacientes con enfisema se han observado una reducción del VEGF y un aumento de la apoptosis de las células endoteliales29. Se podría establecer un círculo vicioso entre la apoptosis y el estrés oxidativo porque las células que entran en apoptosis provocan a su vez un aumento del estrés oxidativo que podría contribuir de nuevo a más apoptosis. En este sentido, se ha descrito que la enzima antioxidante superóxido dismutasa protege frente al desarrollo de la apoptosis y del enfisema inducido por el bloqueo de los receptores del VEGF30. De igual modo, la inhibición de las caspasas reduce la expresión de marcadores de estrés oxidativo en el pulmón30. Estos datos permiten plantear la posibilidad de que varios factores, como la proteólisis excesiva, la apoptosis y el estrés oxidativo, interactúan para destruir el parénquima y provocar enfisema. La desaparición del tejido pulmonar existente en el enfisema podría deberse a la pérdida progresiva de las células epiteliales y endoteliales mediante apoptosis.
Papel de las infecciones
El papel exacto de las infecciones sobre la evolución natural de la enfermedad se desconoce, pero estos datos vienen a confirmar su posible influencia en el mantenimiento tanto de la actividad destructiva pulmonar como en la actividad inflamatoria-reparadora en estas personas, y previsiblemente influye en la evolución funcional de la EPOC. Las infecciones crónicas estimulan un círculo vicioso del proceso inflamatorio con aumento del número de neutrófilos por inducción de citocinas. Se sabe que los pacientes con EPOC presentan colonizaciones bacterianas y frecuentes episodios de exacerbaciones por infecciones; por ello, parece razonable pensar que haya pacientes en los que la inflamación que produce las infecciones recurrentes contribuya al progreso de la enfermedad. En este sentido se ha mostrado que pacientes con EPOC y con presencia de microorganismos patógenos en las vías aéreas tienen aumentadas las concentraciones de neutrófilos, TNF-α e IL-8 y presentan valores menores en el FEV1.
Fuerzas mecánicas
Aunque poco conocida, una de las primeras teorías sobre la patogenia del enfisema se basó en la acción de las fuerzas mecánicas existentes en el pulmón. La idea de que las fuerzas mecánicas estaban involucradas en la progresión de algunas enfermedades pulmonares ya fue descrita por West31 en 1971. La sucesión de los ciclos respiratorios estables que configuran la respiración normal está intermitentemente interrumpida por inspiraciones profundas en las que las fuerzas se incrementan de forma significativa en el tejido7. Por este motivo, esta teoría plantea que las fuerzas mecánicas que se producen durante la respiración podrían dañar las paredes alveolares si éstas han sufrido una lesión previa, como la que puede haber en el proceso de remodelado tisular de la EPOC o la existente en los modelos químicos de enfisema pulmonar por administración de elastasa o cloruro de cadmio. Actualmente, experimentos llevados a cabo en pacientes sometidos a cirugía de reducción de volumen de pulmón han revelado que las fuerzas mecánicas pueden ayudar a la progresión del enfisema. Estos resultados se han corroborado con los obtenidos en modelos animales de enfisema, donde pudo observarse que las fibras nuevas de colágeno y elastina generadas en la remodelación existente tras la instilación de elastasa presentaban distorsiones debidas a las fuerzas mecánicas que se producen durante la respiración32. Más aún, estos autores llegaron a observar que incluso las propias fuerzas mecánicas por sí solas podrían contribuir a la destrucción del tejido y a la progresión del enfisema.
Desde la inflamación a la remodelación
En la EPOC, la obstrucción está principalmente situada en la periferia pulmonar, donde se sitúan las vías aéreas pequeñas, que abarcan desde la cuarta hasta la decimocuarta generación bronquial y tienen menos de 2 mm de diámetro. Esta zona contribuye en menos del 25% a las resistencias totales al flujo aéreo; de ahí que se le denominara la "gran zona silente", puesto que incluso estando muy afectadas apenas llegan a presentar alteración funcional1.
Además de la alteración en la vías aéreas, la pérdida de retracción elástica que se asocia a la remodelación del tejido conectivo peribronquiolar provoca una rotura de las ataduras alveolares constituidas por los septos alveolares conectados con los bronquiolos y que ejercerían una acción de tirante que impide el colapso de la vía aérea (fig. 1). La importancia de la pérdida de estas ataduras se puso de manifiesto al demostrarse una correlación entre el número de ataduras y el valor del FEV113. Las alteraciones vasculares también forman parte de los cambios histológicos de la EPOC, como muestra el hecho de la presencia de linfocitos T (CD8+) en las arterias pulmonares33.

Fig. 1. Ataduras alveolares. Representación esquemática del efecto funcional de las ataduras alveolares como elementos sostenedores de la luz de las vías aéreas (A). Bronquiolo con reducción del número de ataduras alveolares en un pulmón con enfisema panacinar inducido con elastasa en rata (B). Bronquiolo normal de un pulmón sano de rata con sus ataduras alveolares conservadas (C).
El componente de enfisema de la EPOC se puede clasificar, según la forma en que el ácino esté destruido6, en enfisema centroacinar, que se desarrolla en la porción central del ácino, cerca de los bronquiolos respiratorios y predomina en los lóbulos superiores, y en enfisema panacinar, que implica el agrandamiento de los espacios aéreos distribuido de una manera más uniforme a lo largo de todo el ácino, se observa con mayor frecuencia en las regiones pulmonares inferiores y está especialmente relacionado con el déficit de la enzima α1-antitripsina. Actualmente se sabe que ambos tipos de enfisema pueden coexistir en el mismo pulmón y, exceptuando los casos puros de déficit de la enzima α1-antitripsina, no tienen una diferenciación patogénica reconocida en el desarrollo de la EPOC. Se reconoce al tabaco como principal agente causante de esta enfermedad, aunque no se sabe por qué en su desarrollo predomina un tipo u otro de enfisema. Por ello, las nuevas normativas prefieren utilizar este término de EPOC en lugar del de enfisema o bronquitis crónica por separado34. Existe otra variedad anatómica de enfisema, el enfisema distal o paraseptal, que se localiza en regiones subpleurales del pulmón y en los adyacentes a los septos pulmonares. Esta forma puede evolucionar hacia la formación de bullas y neumotórax.
Además de las diferencias en la situación del ácino afectado, las 2 formas morfológicas mayores (centroacinar y panacinar) tienen también distintas características funcionales (fig. 2) y de afectación tanto de las pequeñas vías aéreas como del tejido conectivo35. Respecto a las diferencias funcionales, se han observado una mayor distensibilidad y pérdida de retracción elástica en las formas de predominio panacinar, que tienden a estar conservadas, e incluso disminuidas, en las formas de predominio centroacinar por la presencia de un componente asociado de fibrosis peribronquiolar. Este último tipo de enfisema tiene además un mayor componente de inflamación de las vías aéreas que produce una disminución de los flujos pulmonares más acentuada que la que se observa en el enfisema de predominio panacinar.

Fig. 2. Preparaciones de hematoxilina-eosina (x40) de un pulmón control de rata, un enfisema centroacinar inducido con cloruro de cadmio y un enfisema panacinar inducido con elastasa en ratas. La presencia de fibrosis peribronquiolar en el enfisema centroacinar confiere la diferenciación funcional de una conservación o incluso reducción de la distensibilidad pulmonar y de una mayor reducción de los flujos espiratorios.
Estudiando el tamaño de los espacios aéreos mediante el análisis de la intersección lineal media36, que determina la distancia entre paredes alveolares y, por tanto, el grado de agrandamiento de los espacios aéreos, se observa un aumento en los pacientes con enfisema en relación con los individuos sanos. Bioquímicamente se han comprobado a su vez diferencias en el contenido de colágeno y elastina, con un aumento de colágeno en el enfisema de tipo centroacinar y una disminución significativa de elastina en los enfisemas de tipo panacinar y centroacinar graves35.
Aunque el enfisema se ha relacionado con la degradación de elastina, sería obvio suponer que el resto de los componentes del tejido conectivo de la matriz extracelular estarían igualmente afectados. De hecho, existe un grupo de enzimas de la matriz, las metaloproteasas, que destruirían las fibras de colágeno. Diversos autores han encontrado una relación entre la inflamación y el grosor de todos los compartimientos de la pared16. Este aumento de grosor de la pared también se observó en fumadores con síntomas de obstrucción crónica de las vías aéreas en comparación con fumadores asintomáticos y función pulmonar normal, lo que indica la presencia de una reparación eficaz cuya función sería preservar la estructura básica encargada del proceso del intercambio gaseoso en el parénquima pulmonar37. Cuando la reparación no es eficaz, existe remodelado, los componentes de la matriz se desorganizan, pierden sus características y su distribución anatómica originales y provocan un cambio en las propiedades elásticas tisulares (fig. 3).

Fig. 3. Remodelación de la arquitectura de las fibras de elastina y de colágeno en el enfisema inducido por elastasa en ratas. Las fibras de elastina, que forman una lámina continua, se engrosan y apolillan. Las fibras de colágeno, que forman haces alrededor de los espacios aéreos, también se hipertrofian de manera significativa. (Datos propios.)
El principal componente de la matriz extracelular es el colágeno, vital para mantener la estructura pulmonar. Los colágeno de tipos I y III están presentes en la capa adventicia de las arterias pulmonares, en el intersticio del árbol bronquial, en el septo interlobular, en la lámina propia bronquial y en el intersticio alveolar, lugares donde ocurren todos los cambios en el enfisema. Varios autores han encontrado una asociación entre el enfisema y evidencias morfométricas de rotura y reparación de colágeno38. Los diversos resultados respecto al contenido de colágeno en el enfisema reflejan que el remodelado de la matriz es un proceso dinámico con degradación de colágeno, seguido por un proceso de reparación que lleva a un incremento de la deposición del mismo. La heterogeneidad de la lesión enfisematosa y su distribución parcheada en el pulmón, así como los diferentes métodos utilizados, hacen difícil la determinación del contenido proteico. Estudios bioquímicos han demostrado la existencia de un aumento del contenido de colágeno tanto en pacientes con enfisema2,35 como en modelos animales de enfisema39 (fig. 4). Estudios llevados a cabo con microscopia electrónica también han demostrado la existencia de procesos de destrucción y reparación de las fibras de colágeno39. Este incremento de colágeno no significa que la enfermedad se acompañe de fibrosis, pero su presencia, aparentemente paradójica en un sistema que se define como con pérdida de tejido, reflejaría un fracaso de los sistemas de reparación que siguen a la lesión y podría justificar que se planteasen medidas de tratamiento antifibrótico experimental como paso previo a una posible nueva línea terapéutica dirigida a la regeneración pulmonar40.

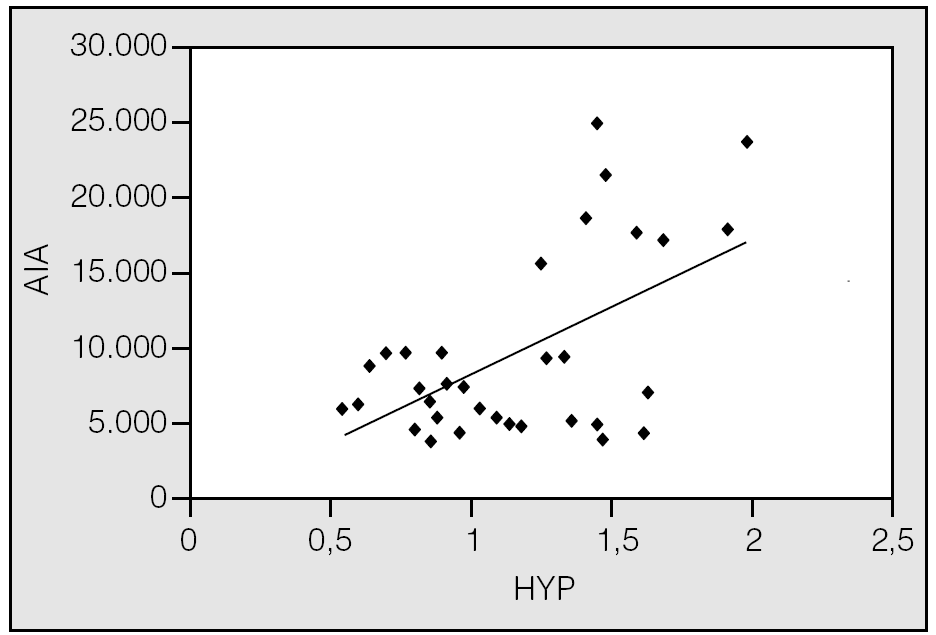
Fig. 4. Correlación positiva entre el tamaño de los espacios aéreos, representado por el área interior alveolar (AIA), y el contenido de colágeno cuantificado como hidroxiprolina (HYP) en ratas controles y con enfisema inducido con elastasa43.
El principal componente de las fibras elásticas es la elastina, material proteico muy insoluble que contiene 2 aminoácidos exclusivos, la desmosina y la isodesmosina. Las fibras elásticas son estructuras muy estables responsables de la capacidad de la retracción elástica del pulmón. Existen muchas evidencias que indican que la elastina está involucrada en el enfisema pulmonar. Hay una relación entre la gravedad de la enfermedad y la pérdida de elastina en pulmones con enfisema, y también entre la expresión de ARN mensajero de elastina y el tamaño medio de los espacios aéreos distales, lo que indicaría la existencia de un proceso de reparación41.
En pacientes con EPOC se ha encontrado que el contenido de elastina puede estar disminuido en todos los tipos de enfisema28, aunque otros autores lo han encontrado aumentado38. En modelos experimentales de enfisema, el contenido de elastina se ha encontrado aumentado en un modelo de hámsters tratados con cloruro de cadmio50 o en valores normales, como en el caso del enfisema pulmonar inducido con elastasa39.
Estos procesos de reparación pueden ser bioquímicamente efectivos, es decir, puede que presenten una cuantificación normal o alta, pero morfológicamente defectuosos, sin seguir una distribución arquitectónica regular, con pérdida de la alineación natural de la elastina, lo que provocaría que la acción de enzimas elastolíticas pueda sea mayor en estas fibras defectuosas y que las fuerzas mecánicas del pulmón puedan romperlas con mayor facilidad.
Desde la remodelación a los cambios mecánicos
Durante la respiración tranquila a volumen corriente los alveolos permanecen en todo momento comunicados con el exterior gracias a una vía aérea que se mantiene abierta por su soporte cartilaginoso en las primeras generaciones o por los haces de fibras conectivas y soporte traccional del parénquima en las últimas generaciones. Sin embargo, puede producirse un cierre temprano de estas vías, dejando aire alveolar atrapado, durante una maniobra de espiración forzada. Se produce un colapso de las vías periféricas porque las fuerzas de tracción y de los haces del tejido conectivo que tienden a mantenerlas abiertas van disminuyendo a medida que el pulmón se deflaciona y se ven sobrepasadas por la elevada presión intrapleural que provoca la espiración forzada. En personas normales este fenómeno sucede próximo al volumen residual, pero en pacientes con EPOC el cierre se anticipa y también puede aparecer durante la respiración a volumen corriente. Los cambios descritos en las vías aéreas y el parénquima favorecen este cierre temprano, que no es homogéneo en el árbol bronquial, ya que presenta un elevado grado de heterogeneidad a lo largo de las ramificaciones de la vía aérea. Los mecanismos fisiológicos o fisiopatológicos de esta heterogeneidad se desconocen, pero pueden producir descensos de flujo en unas partes e incrementos en otras; irregularidades que deben de tener impacto en la función pulmonar, pero que no pueden detectarse por las pruebas habituales de función pulmonar42. Las curvas de presión-volumen o de flujo-volumen del pulmón proporcionan una información valiosa pero limitada porque dan por supuesto un comportamiento homogéneo del pulmón, algo que realmente no sucede43. Aparte de los estudios fisiológicos de ventilación base-vértice, dependientes de las fuerzas de la gravedad, las primeras evidencias de la existencia de heterogeneidades de expansión del parénquima se obtuvieron mediante aplicación de cápsulas pleurales para medir las presiones alveolares44 o de marcadores radioopacos para examen fluoroscópico45. Ambas técnicas mostraron la existencia de expansión y deflación pulmonares heterogéneas, más allá de las propias debidas al efecto gravitacional base-vértice. Las técnicas de imagen han permitido profundizar en esta heterogeneidad de las vías aéreas en sus movimientos de expansión-deflación y en la aparición del atrapamiento aéreo. Mediante la tomografía computarizada (TC) de alta resolución en perros anestesiados se ha podido demostrar que incluso en condiciones normales existe una variabilidad en el diámetro de las vías aéreas de unos días a otros. Si estos perros eran expuestos a histamina, intravenosa o en aerosol, la respuesta era muy irregular de unas vías aéreas a otras. Lo mismo sucedía en pacientes tras ser expuestos a metacolina42. La utilización de la TC con cortes en inspiración y espiración también permite observar las zonas de atrapamiento aéreo del pulmón46. La resonancia magnética con gases nobles hiperpolarizados puede aportar mayor definición que la TC, aunque por ahora de una manera muy experimental. Mediante la inhalación de helio-3 hiperpolarizado pueden realizarse adquisiciones dinámicas muy rápidas que permiten construir un mapa completo de tamaños alveolares47 (fig. 5) y de las velocidades de acceso del gas a cada unidad, tiempos de llenados y tasas de insuflación durante una inspiración, con una definición aproximada48 de 1 mm2.

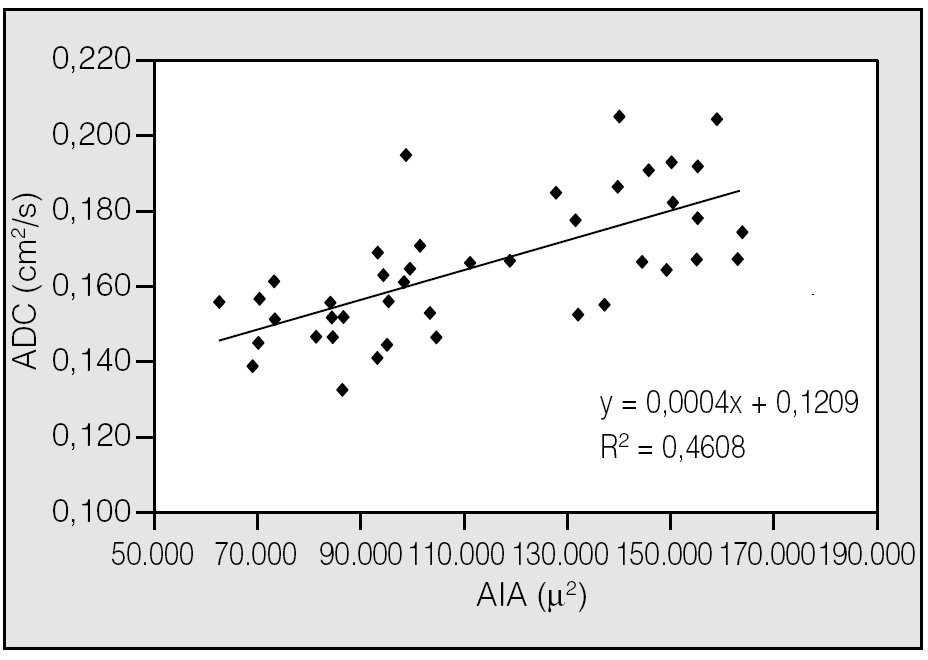
Fig. 5. Correlación significativa positiva entre la medida del área alveolar interna (AIA) y los valores de coeficientes aparentes de difusión (ADC) estimados píxel a píxel mediante la aplicación de la resonancia magnética con gases hiperpolarizados en el estudio de la función pulmonar en ratas normales y con enfisema inducido con elastasa en grados leves de enfisema, donde la resolución de dicha técnica sigue siendo por ahora insuficiente para detectarlos52.
Las pruebas mecánicas de función pulmonar (espirometría o pletismografía) promedian necesariamente la existencia de estas heterogeneidades, por lo que pueden arrojar una función falsamente normal. Si existe atrapamiento aéreo regional o heterogéneo, unas pruebas funcionales normales tendrán un valor muy limitado y sólo cuando estas lesiones se agravan o se diseminan podrán detectarse funcionalmente. En los primeros casos, las alteraciones encontradas posiblemente no tendrán importancia clínica, pero pueden ser la única evidencia de que se está presentando una enfermedad de las vías aéreas.
El atrapamiento aéreo se produce siempre que exista un cierre temprano de las vías aéreas durante la espiración. La hiperinsuflación dinámica se produce por cambios agudos en el cierre temprano de estas vías y siempre que el tiempo espiratorio sea insuficiente para alcanzar el punto de reposo previo al siguiente ciclo respiratorio. En las situaciones acompañadas de un incremento de la demanda ventilatoria, como el ejercicio, y en los casos de exacerbación, este fenómeno se agrava, se rompe el equilibrio antes alcanzado y se produce hiperinsuflación dinámica y autopresión positiva al final de la espiración (PEEP), con el consiguiente empeoramiento funcional añadido (fig. 6). También puede suceder que se produzca auto-PEEP e hiperinsuflación dinámica en unas unidades pulmonares y no en otras, con un resultado global que no altere las medidas pulmonares totales. Estos fenómenos de irregularidades en los cierres de unas unidades respecto de otras tienen de por sí una tendencia a la distribución interregional, con aparición más intensa y temprana en las zonas pulmonares dependientes y con intensificación si se realizan espiraciones forzadas. También es posible lo contrario, presencia de auto-PEEP sin atrapamiento acompañante cuando la activación de los músculos espiratorios desplaza el volumen de reposo, como sucede en sujetos sanos durante el ejercicio o en respuesta a una PEEP extrínseca y en pacientes con EPOC por incremento de presión abdominal producido por la acción de los músculos abdominales durante la espiración. Aunque la obstrucción al flujo aéreo es la principal causa de atrapamiento aéreo, su presencia no predice el grado real de hiperinsuflación. De hecho, si los flujos espiratorios disponibles en un sujeto son suficientes para mantener una adecuada ventilación en reposo, el volumen teleespiratorio no tiene por qué aumentar, aunque exista limitación al flujo aéreo49. En todos los casos de EPOC el concepto de obstrucción al flujo aéreo queda implícito por la presencia de una espirometría obstructiva, espirometría que debe realizarse con el máximo esfuerzo espiratorio; sin embargo, este hecho no implica que durante una respiración a volumen corriente exista también una limitación al flujo aéreo. Para distinguir estos 2 conceptos, algunos autores utilizan el término de "limitación al flujo espiratorio" cuando ésta también se presenta en la respiración a volumen corriente, porque no todos los casos de EPOC tienen también limitación al flujo espiratorio a volumen corriente50. Cuando la limitación al flujo espiratorio llega a presentarse durante la respiración a volumen corriente, aparecerán los fenómenos de hiperinsuflación dinámica y de auto-PEEP, con el consiguiente incremento en el trabajo de la respiración, desventaja mecánica de los músculos inspiratorios, consecuencias hemodinámicas y aparición de la disnea.

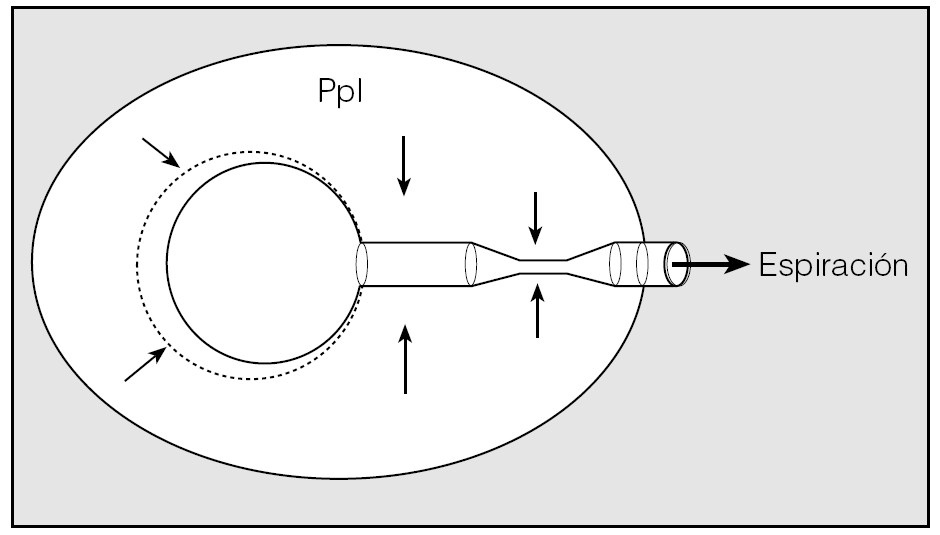
Fig. 6. Esquema de funcionamiento mecánico del sistema respiratorio. Durante la inspiración, la presión pleural (Ppl) negativa se transmite a todas las estructuras intratorácicas y se facilita la expansión de las vías aéreas y alveolos. Durante la espiración, la Ppl positiva, igualmente transmitida a todas las estructuras intratorácicas, puede provocar el colapso de la vía aérea cuando la presión de su interior se iguala a la pleural. En las enfermedades obstructivas, al acortar el tiempo espiratorio este colapso se acentúa y se anticipa el atrapamiento aéreo.
A pesar de las importantes consecuencias que tiene la presencia de la limitación al flujo espiratorio en los pacientes con EPOC, su prevalencia y significado clínico no se han estudiado adecuadamente hasta la actualidad.
Conclusión
La EPOC se inicia con la exposición al humo del tabaco y el desarrollo de una respuesta inflamatoria característica que afecta a las vías aéreas, el parénquima y las arterias pulmonares. Cuando esta inflamación se sigue del remodelado característico, quizá se pueda empezar a hablar de que la enfermedad ya está instaurada, aunque el componente de obstrucción, imprescindible para su diagnóstico, no pueda detectarse en estas primeras fases de la enfermedad. Esto no quiere decir que no pueda ya existir, sino que simplemente la limitación del FEV1 para detectar lesiones leves lo impide. Además, en su inicio la lesión es muy heterogénea, con afectación posiblemente parcheada que combina zonas patológicas con otras más respetadas que no son capaces de transmitir una anomalía global detectable en las pruebas funcionales. El atrapamiento aéreo sigue este mismo patrón heterogéneo, que en sus primeras fases podría detectarse mediante pruebas de imagen y posiblemente, sólo cuando es difuso, en las pruebas funcionales habituales. Queda por delante un futuro próximo abierto a un nuevo concepto de diagnóstico por imagen funcional cuyas expectativas son prometedoras, pero que aún está por desarrollar.
Correspondencia: Dr. G. Peces-Barba Romero.
Servicio de Neumología. Fundación Jiménez Díaz-UTE.
Avda. Reyes Católicos, 2. 28040 Madrid. España.
Correo electrónico: gpecesba@separ.es







