
La alfa-1 antitripsina (AAT) es el inhibidor de proteasas más abundante del suero humano con concentraciones promedio en individuos sanos que oscilan entre 120-220mg/dl determinadas por nefelometría y cuya síntesis depende del gen SERPINA11,2.
Los alelos deficitarios más frecuentes encontrados en España son el PI*S y el PI*Z, con una tasa de portadores aproximada en población general española de 1/5 y 1/61 sujetos, respectivamente3-5, y cuya existencia se ha relacionado con el desarrollo de enfisema pulmonar en periodos precoces de la edad adulta, hepatopatías en niños y adultos, así como con otras entidades como son las vasculitis sistémicas (especialmente las granulomatosis de Wegener c-ANCA positivas) o paniculitis necrosante2,6.
En Canarias existen pocos estudios genéticos poblacionales de alelos deficitarios de ATT7, y ninguno que identifique variantes deficitarias infrecuentes. Por este motivo, el objetivo de este trabajo es describir el espectro de mutaciones encontradas en la población de Tenerife atendida en el Hospital Universitario Nuestra Señora de Candelaria (HUNSC).
Estudio transversal y descriptivo de una muestra que comprende a casos-índice con niveles plasmáticos de AAT inferiores a 100mg/dl que fueron remitidos al servicio de Análisis Clínicos del HUNSC por sospecha clínica de déficit de alfa-1-antitripsina (DAAT) durante el periodo comprendido entre el 1 de enero de 2009 y el 31 de agosto de 2016.
La determinación de los valores séricos de AAT se realizó mediante nefelometría (BN ProSpec System, Siemens). El ADN genómico fue purificado de sangre total EDTA (QIAamp DNA Blood Mini Kit, Qiagen). Los dos alelos deficitarios más frecuentes PI*S y PI*Z fueron genotipados por PCR a tiempo real con sondas FRET (LightCycler 2.0). Los genotipos obtenidos fueron correlacionados con los valores de AAT plasmáticos detectados previamente siguiendo el protocolo establecido en nuestro hospital8. A los pacientes con niveles plasmáticos de ATT discordantes con los esperados para los genotipos S/Z obtenidos se les realizó la secuenciación completa de los exones codificantes y las zonas intrónicas flanqueantes del gen SERPINA1 (BigDye v3.1, Thermo Fisher), sustituyendo la realización de prueba clásica del fenotipo por isoelectroenfoque. La secuenciación de SERPINA1 fue realizada en un secuenciador capilar AB3500 (Applied Biosystems), comparando los resultados obtenidos con la secuencia de referencia NM_001127701.1 (SeqScape 3.0).
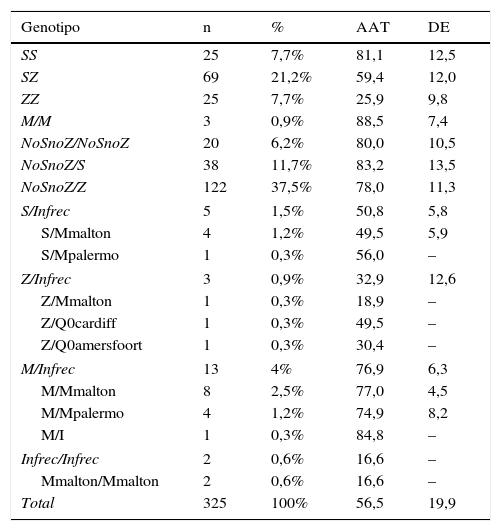
En el estudio fueron incluidos 325 pacientes con DAAT que presentaron niveles plasmáticos de AAT<100mg/dl. A todos los pacientes se les realizó el estudio dirigido a alelos deficitarios PI*S y PI*Z. Los genotipos obtenidos se muestran en la tabla 1.
Genotipos y niveles de AAT obtenidos en el estudio
| Genotipo | n | % | AAT | DE |
|---|---|---|---|---|
| SS | 25 | 7,7% | 81,1 | 12,5 |
| SZ | 69 | 21,2% | 59,4 | 12,0 |
| ZZ | 25 | 7,7% | 25,9 | 9,8 |
| M/M | 3 | 0,9% | 88,5 | 7,4 |
| NoSnoZ/NoSnoZ | 20 | 6,2% | 80,0 | 10,5 |
| NoSnoZ/S | 38 | 11,7% | 83,2 | 13,5 |
| NoSnoZ/Z | 122 | 37,5% | 78,0 | 11,3 |
| S/Infrec | 5 | 1,5% | 50,8 | 5,8 |
| S/Mmalton | 4 | 1,2% | 49,5 | 5,9 |
| S/Mpalermo | 1 | 0,3% | 56,0 | – |
| Z/Infrec | 3 | 0,9% | 32,9 | 12,6 |
| Z/Mmalton | 1 | 0,3% | 18,9 | – |
| Z/Q0cardiff | 1 | 0,3% | 49,5 | – |
| Z/Q0amersfoort | 1 | 0,3% | 30,4 | – |
| M/Infrec | 13 | 4% | 76,9 | 6,3 |
| M/Mmalton | 8 | 2,5% | 77,0 | 4,5 |
| M/Mpalermo | 4 | 1,2% | 74,9 | 8,2 |
| M/I | 1 | 0,3% | 84,8 | – |
| Infrec/Infrec | 2 | 0,6% | 16,6 | – |
| Mmalton/Mmalton | 2 | 0,6% | 16,6 | – |
| Total | 325 | 100% | 56,5 | 19,9 |
AAT: alfa-1 antitripsina; DE: desviación estándar; Infrec: variantes deficitarias infrecuentes.
Se detectaron 46 pacientes (14,2%) con un genotipo asociado a un déficit grave (AAT<50mg/dl), de los cuales 24 fueron PI*ZZ, 10 PI*SZ, uno PI*SS, 2 PI*S/Mmalton, uno PI*Z/Q0amersfoort, uno PI*Z/Q0cardiff, uno PI*Z/Mmalton, 2 PI*Mmalton/Mmalton, uno PI*MZ, 2 PI*S/NoSnoZ y uno PI*Z/NoSnoZ. Con déficit leve-moderado (AAT entre 50 y 100mg/dl) se registraron 279 pacientes (85,8%), distribuidos de la siguiente manera: 119 PI*Z/NoSnoZ, 59 PI*SZ, 36 PI*S/NoSnoZ, 24 PI*SS, 8 PI*M/Mmalton, 4 PI*M/Mpalermo, 2 PI*S/Mmalton, uno PI*S/Mpalermo, uno PI*ZZ, uno PI*MZ, uno PI*MI, 3 PI*MM y 20 NoSnoZ/NoSnoZ.
El porcentaje de pacientes portadores de alelos deficitarios infrecuentes fue del 7,7%, correspondiendo el 80% de estos a pacientes con la mutación F76del (PI*Mmalton y PI*Mpalermo).
En este estudio hemos realizado un protocolo de diagnóstico de DAAT basado en la cuantificación de los niveles plasmáticos de AAT seguido del genotipo dirigido a alelos deficitarios PI*S y PI*Z por ser los más frecuentes en nuestra población. La correlación de ambos resultados ha demostrado en los últimos años ser la técnica más eficiente, consiguiendo un diagnóstico inequívoco en aproximadamente el 96% de los casos8. En el resto de pacientes se realizó la secuenciación completa del gen SERPINA1, técnica que ha demostrado mejorar la precisión en el diagnóstico de DAAT9. En la mayoría de protocolos de estudio de DAAT se suele aplicar como test de referencia el fenotipo mediante isoelectroenfoque. Sin embargo, en nuestro caso se decidió realizar la secuenciación del gen SERPINA1 debido a las limitaciones que presenta el fenotipado en la identificación de las variantes deficitarias infrecuentes10.
Un hallazgo destacable en nuestro estudio es el rendimiento obtenido al aplicar la combinación de niveles proteicos y genotipo de alelos PI*S y PI*Z (84,9%), siendo inferior a lo que se describe en la literatura8. Este bajo rendimiento del protocolo diagnóstico es consecuencia de la alta prevalencia de mutaciones deficitarias infrecuentes detectadas, especialmente la mutación F76del (PI*Mmalton y PI*Mpalermo), que supone el 81,5% de las variantes infrecuentes encontradas, porcentaje superior a lo descrito por estudios similares realizados en España11. Adicionalmente, en los 25 pacientes a los que se realizó la secuenciación del gen SERPINA1 se encontraron 20 pacientes NoSnoZ/NoSnoZ, 4 pacientes PI*S/NoSnoZ y un paciente con genotipo PI*Z/NoSnoZ con niveles plasmáticos de ATT discordantes con el genotipo dirigido S/Z, a los cuales, siendo susceptibles de secuenciación completa de SERPINA1, no se les realizó la técnica al no encontrarse disponible en nuestro hospital en el momento del estudio. Debemos añadir que en 3 pacientes con déficit leve-moderado de AAT no se han encontrado mutaciones a pesar de realizar la secuenciación completa de SERPINA1, lo cual pudiera ser debido a mutaciones en regiones reguladoras del gen no estudiadas12 o mutaciones no detectables por secuenciación que precisan técnicas alternativas para su diagnóstico13.
Debido a que el DAAT es una de las enfermedades hereditarias con más prevalencia en nuestra población y a que la mutación F76del, al igual que el alelo PI*Z, ha sido asociada al desarrollo de enfisema pulmonar y enfermedad hepática14,15, creemos conveniente valorar en nuestra población su detección en todos aquellos pacientes en los que no se consigue hacer un diagnóstico inequívoco con los protocolos genéticos tradicionales que combinan medida de AAT sérica, el genotipado dirigido a los alelos deficitarios PI*S y PI*Z, y el fenotipo.







