

La hipertensión pulmonar (HTP) es una enfermedad heterogénea que se caracteriza por una presión de la arteria pulmonar mayor de 20 mmHg en reposo o 30 mmHg durante el ejercicio, que generalmente se debe a un aumento de la resistencia vascular pulmonar y en sus últimos estadios cursa con insuficiencia cardíaca. Existen una variedad primaria, cuyo origen no ha sido completamente caracterizado, y una variedad secundaria a diversas enfermedades como la tromboembolia crónica, enfermedades del colágeno vascular, cardiovalvulopatías izquierdas, vasoconstricción secundaria a hipoxia, infección por el virus de la inmunodeficiencia humana y fármacos (anorexígenos), entre otras1. Todos estos tipos de HTP, tanto de la variedad primaria como secundaria, cursan con manifestaciones clínicas similares.
En cuanto a la HTP primaria (HTPP), se ha establecido que por cada millón de personas una padece esta enfermedad, con una mayor prevalencia en el sexo femenino (1,7:1) comparado con el masculino2. Entre los casos de HTPP muchos se presentan en la misma familia, con un carácter hereditario, en lo que se conoce como HTPP familiar. Otros casos se califican de esporádicos, sin que en ellos se evidencien antecedentes familiares de HTPP; es lo que se denomina HTPP no familiar o esporádica. Cuando la HTPP se diagnostica, generalmente se encuentra en sus fases más avanzadas debido a la inespecificidad de los síntomas en la fase leve, por lo cual se desconoce en detalle la historia natural de esta enfermedad. Dependiendo de la clase funcional de la New York Heart Association en la que se encuentren, los pacientes tienen una supervivencia de 6 meses a 3,5 años desde el momento en que se establece el diagnóstico, y la gran mayoría fallece debido a insuficiencia cardíaca y muerte súbita2.
La fisiopatología depende en gran medida del carácter primario o secundario de la HTP. Tradicionalmente se ha establecido que la HTPP se desarrolla debido a una alteración, hasta ahora desconocida, que conduce a un desequilibrio entre los agentes vasoactivos que influyen en el músculo liso del lecho vascular pulmonar, con predominio de vasoconstricción, cuya consecuencia es un aumento de la resistencia vascular, y por ende de la presión arterial pulmonar, que a largo plazo genera cambios morfológicos en la pared de las arterias (fig. 1). Es bien sabido que el endotelio es la estructura fundamental que produce estos agentes vasoactivos que actúan sobre la capa media de las arterias, por lo que cualquier alteración de estos mediadores debe de ser producto de una alteración en la función endotelial. El remodelado vascular en la HTP, sea ésta de origen primario o secundario, es casi idéntico y se caracteriza por la proliferación de la capa media y el engrosamiento de la íntima, con formación de lesiones plexiformes, las cuales constituyen una masa de vasos desorganizados, con células endoteliales proliferantes, miofibroblastos y células de músculo liso, que en la HTPP se presentan en mayor proporción en las pequeñas arterias y arteriolas, junto con el engrosamiento obstructivo de la íntima2,3. En la variedad primaria, las lesiones plexiformes y concéntricas están compuestas por una población de células endoteliales monoclonales; en cambio, en la secundaria no es así4. Este hecho indicaría que podría haber una alteración genética en estas células que causara el avance de su crecimiento en la HTPP; estas células endoteliales defectuosas serían responsables de las alteraciones del tono vascular y del desarrollo de todo el complejo histopatológico que caracteriza la HTPP, mediado por señales humorales. Sin embargo, es posible que estas lesiones representen un cambio adaptativo erróneo del lecho vascular inducido por el aumento de la presión.
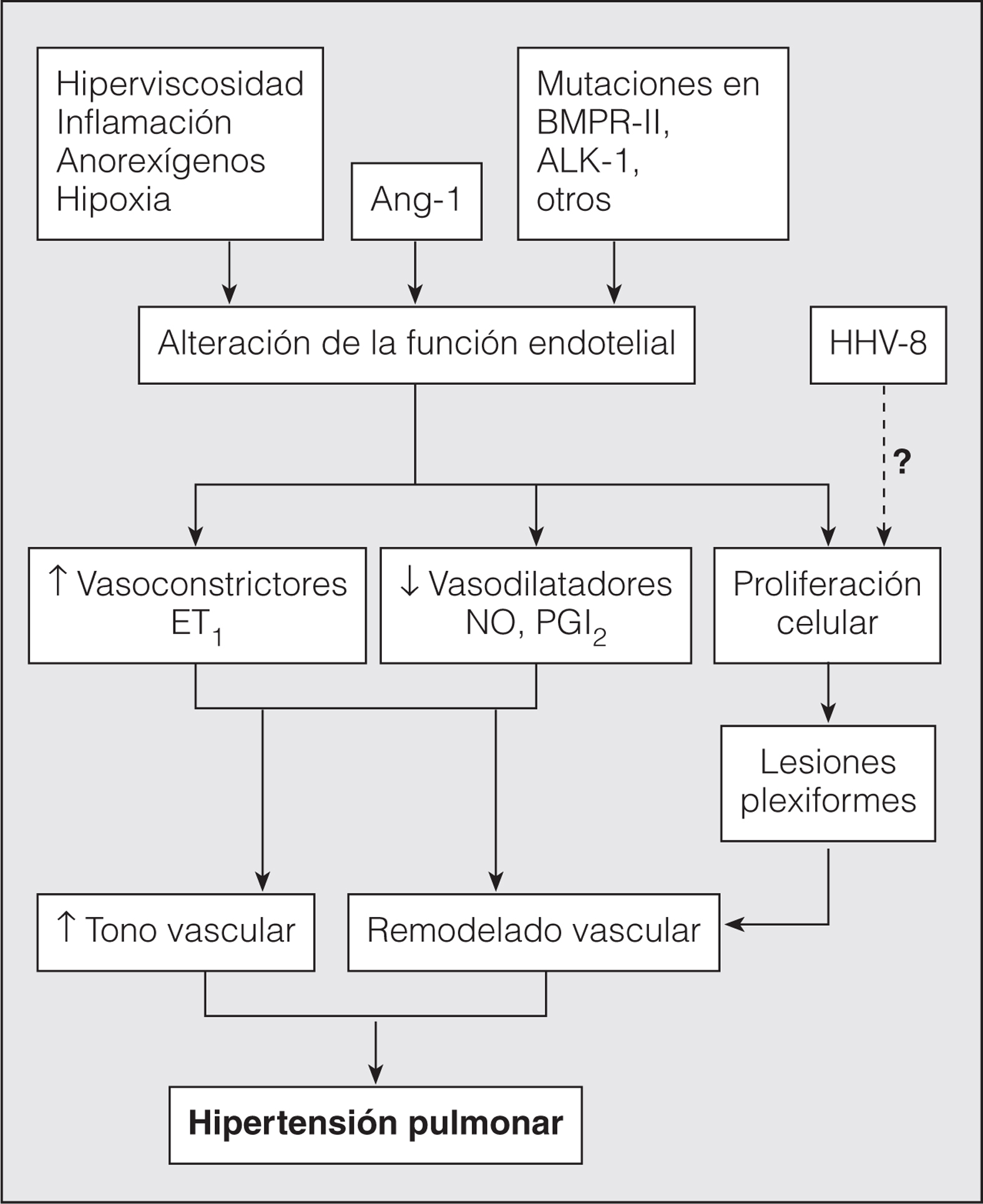
Fig. 1. Esquema de la fisiopatología de la hipertensión pulmonar. La función inadecuada del endotelio y el desequilibrio resultante de las sustancias vasoactivas que éste genera son el proceso central en la fisiopatología de la hipertensión pulmonar primaria. Ang-1: angipoyetina 1; ET1: endotelina 1; NO: óxido nítrico; PGI2: prostaciclina; HHV-8: virus herpes humano tipo 8.
Se ha estudiado ampliamente la genética en la HTPP familiar y se ha observado un patrón hereditario autosómico dominante. La enfermedad se manifiesta en un 10-20% de los individuos portadores de la mutación y, además, cada nueva generación que posee la mutación manifiesta la enfermedad a edades más tempranas, cumpliendo con el fenómeno de anticipación genética5. Se ha localizado la mutación responsable de muchos casos de HTPP familiar en el locus 2q31-q326,7, en el gen que codifica el receptor tipo II de las proteínas morfogénicas del hueso (BMPR-II)8,9, cuyo ligando son las proteínas de la superfamilia del factor de crecimiento transformador tipo beta (TGF-β), entre las que se encuentra, evidentemente, el TGF-β, así como las proteínas morfogénicas del hueso (BMP). Estos factores de crecimiento regulan la diferenciación y apoptosis de muchas líneas celulares, entre ellas el músculo liso vascular, fundamentalmente inhibiendo su crecimiento y proliferación. Por lo tanto, una disfunción del BMPR-II abatiría la señal antiproliferativa de las proteínas de la superfamilia del TGF-β sobre el músculo liso vascular del lecho pulmonar, lo que contribuiría a las alteraciones de la capa media observadas en HTPP10. También se han descrito mutaciones en el gen ALK-1, el cual codifica el receptor tipo I del TGF-β en individuos con HTP y telangiectasia hemorrágica hereditaria11. No obstante, hay muchos casos de HTPP familiar en los que no se han encontrado mutaciones en estos 2 genes, lo cual apuntaría a la existencia de otros mecanismos que probablemente involucren mutaciones de genes asociados a la superfamilia del TGF-β, como las Smads, proteínas responsables de la señalización intracelular del BMPR-II. Es probable que estas mutaciones, y muchas otras aún no caracterizadas, no sólo afecten directamente la respuesta del músculo a factores de crecimiento como el TGF-β, sino que también lo hagan más susceptible a las señales paracrinas de un endotelio con alteraciones que pueden deberse en gran parte a defectos genéticos, lo cual desencadenaría una serie de acontecimientos que culminarían en el establecimiento de la HTP y el remodelado vascular.
Uno de los vasomoduladores de mayor importancia involucrados en la HTP es la endotelina 1 (ET1), que es el vasoconstrictor más potente que se conoce hasta la fecha12. La ET1 es un péptido de 21 aminoácidos producido por el endotelio que ejerce su función a través de sus receptores ETA y ETB y de la activación de canales de Ca2+ dependientes de voltaje. En condiciones normales, la ET1 se secreta en cantidades mínimas en la vasculatura pulmonar. Es interesante destacar que en pacientes con HTP la producción está claramente aumentada13 y se correlaciona directamente con la gravedad de la enfermedad y el grado de desarrollo de las lesiones plexiformes; es decir, una alteración en la función endotelial generaría una mayor cantidad de ET1, la cual es responsable en gran medida del aumento de la resistencia vascular pulmonar, al actuar como el potente vasoconstrictor que es. Por otra parte, la ET1 también se ha relacionado con la inducción del remodelado vascular, ya que aumenta la expresión del receptor 1b de la serotonina en la célula de músculo liso vascular14. De ello da fe el tratamiento con antagonistas de los receptores de la ET1 como el bosentán y el sixtasentan, que prometen grandes beneficios. Trabajos que han estudiado a grupos de pacientes con HTP en diferentes clases funcionales de la New York Heart Association, sometidos a tratamiento con bosentán a corto y largo plazo, han dado resultados muy alentadores, con una mejora clínica evidente de los pacientes y una aparente remisión leve del remodelado vascular15-17, lo cual demostraría la importancia que tiene este péptido en la fisiopatología de la hipertensión y, al parecer, también en el proceso de remodelado vascular.
Diversos trabajos han tratado de relacionar ciertos acontecimientos fisiopatológicos comunes a la mayoría de los pacientes con HTP no familiar en un esquema básico de cómo podría desarrollarse la enfermedad18,19. La angiopoyetina 1 (Ang-1), una hormona secretada durante el desarrollo embrionario vascular que no se detecta normalmente en la vida adulta, se encuentra aumentada en la sangre de pacientes con HTPP, y se sabe que su grado de aumento es directamente proporcional a la gravedad clínica de la enfermedad. Estos mismos pacientes presentan una fosforilación aumentada del receptor TIE-2, un receptor de la Ang-1, exclusivo de células endoteliales, y una expresión disminuida del receptor BMPR1A; cabe destacar que pacientes con la forma familiar también tienen disminuida la expresión de este último debido a que el receptor BMPR-II defectuoso tiende a dimerizarse con el BMPR1A. Se ha demostrado in vitro que éstos son efectos causados directamente por la Ang-1, e in vivo Ang-1 induce un aumento de la musculatura de las arteriolas pulmonares19. A través de la señalización de TIE-2, la Ang-1 promueve la liberación endotelial de serotonina, un potente estimulador del crecimiento de las células musculares lisas que forman la capa media de los vasos sanguíneos; dicho crecimiento no se observa con el bloqueo de sus receptores. La serotonina se encuentra aumentada en las capas íntima y media de las arterias pulmonares humanas de pacientes con HTP, pero no en sujetos sanos, en quienes la expresión es muy baja. Estos hallazgos inducen a pensar que algún defecto en la producción de Ang-1 podría ser uno de los acontecimientos fisiopatológicos de gran importancia en la HTP, ya sea un defecto constitutivo o desencadenado por factores externos, o tal vez por una interacción de ambos.
Por otro lado, el óxido nítrico (NO) es otro de los factores vasomoduladores sintetizados por el endotelio que se relacionan con la fisiopatología de la HTP, dado su importante papel como vasodilatador. Es sintetizado a partir de la L-arginina por la NO sintasa, de la cual existen 3 isoformas: una expresada por los macrófagos, otra por las neuronas y, por último, la endotelial. La sintasa endotelial de NO (eNOS) es una isoforma dependiente del calcio, está ampliamente expresada en el endotelio pulmonar de personas sanas, su acción se ha implicado en el mantenimiento de la baja resistencia del circuito vascular pulmonar20 y su metabolismo está claramente alterado en personas con HTP. Su efecto se ejerce a través del guanosinmonofosfato cíclico y mediante la regulación de canales de potasio, por lo que el sildenafilo, un inhibidor de la fosfodiesterasa tipo 5 del guanosinmonofosfato cíclico, ha resultado beneficioso en el tratamiento de la HTP y hasta se ha utilizado como sustituto de la prostaciclina subcutánea (treprostinil) en el tratamiento de un caso de HTP asociada a lupus eritematoso21. En pacientes con HTP se observa una alteración del metabolismo de esta molécula, dado que se ha demostrado que el tejido pulmonar de individuos enfermos muestra una disminución de la expresión de la eNOS, y por consiguiente una disminución de la producción de NO, que se correlaciona directamente con el grado de resistencia vascular y el estadio clínico de la enfermedad22; esto apuntaría a que el principal factor vasodilatador disminuido en la HTP podría ser el NO.
Lo anterior se considera tradicionalmente correcto en relación con el NO y la HTP; sin embargo, evidencias recientes sostienen que las lesiones plexiformes tienen una expresión aumentada de la eNOS, lo que ha suscitado la hipótesis de un posible mecanismo compensador del aumento del tono vascular en la HTP. Paradójicamente este aumento de la expresión de la enzima no siempre se acompaña de un aumento concomitante de la producción de NO23. A su vez, la enzima sintasa de NO inducible, que no se había estudiado antes, demostró estar aumentada en las lesiones plexiformes de pacientes con HTP, probablemente en relación con las fuerzas de cizallamiento. Las limitaciones de este estudio se encuentran en que los métodos de inmunohistoquímica pueden demostrar el aumento de expresión de una proteína, pero no su grado de actividad, de modo que no queda claro si el endotelio estaría expresando una enzima defectuosa, probablemente debido a una mutación, que desencadenaría un defecto en la producción por parte del endotelio, y por consiguiente un efecto deficiente sobre la capa media de los vasos, lo que permitiría el predominio de factores vasoconstrictores como la ET1.
Otro importante producto vasomodulador producido por el endotelio vascular que podría estar afectado en un endotelio disfuncional es la prostaciclina, que también se ha estudiado en la fisiopatología de la HTP. La prostaciclina es el principal metabolito derivado del ácido araquidónico liberado por el endotelio y posee efectos vasodilatadores y como antiagregante plaquetario. En pulmones normales la sintasa de prostaciclina se expresa en mayor cantidad en los vasos pulmonares de mayor calibre, y en menor cantidad en las arteriolas y los vasos de pequeño calibre. Esta característica normal parece estar exagerada en pacientes que padecen HTP, probablemente debido a que es en estas arteriolas donde se presentan en mayor proporción las lesiones plexiformes y el mayor grado de alteración del endotelio; esto tiene como efecto una mayor vasoconstricción y tendencia a la coagulación dentro de estos vasos24, lo que favorece la tromboembolia crónica presente en algunos pacientes con HTP. El tratamiento con prostaciclina (epoprostenol) ha demostrado ser el más eficaz actualmente para el manejo de los pacientes, en especial los que se encuentran en estadios de alto riesgo. El iloprost, un análogo estable de la prostaciclina, se ha usado, administrado por vía inhalatoria, en el tratamiento de pacientes con HTP secundaria a tromboembolia crónica que rechazan la cirugía, y con él se obtienen mejores resultados que con el tratamiento convencional25, no sólo por su efecto vasodilatador, sino también por su efecto antiagregante.
Otro campo de investigación en la HTP son los canales de potasio. Se ha comparado la funcionalidad de éstos en células de músculo liso de la arteria pulmonar de pacientes con HTPP e HTP secundaria, y se ha observado que, comparadas con las de la HTP secundaria, las células de pacientes con la variedad primaria tenían una función reducida de los canales de potasio. Lo anterior apunta a que el músculo liso tendría una mayor respuesta a las señales vasoconstrictoras provenientes del endotelio, debido a que se generan un potencial de membrana más alto y una mayor concentración de calcio citoplásmico; este último tiene un papel central en la vasoconstricción y posiblemente sea un estímulo para la hiperplasia e hipertrofia del músculo liso vascular en la HTPP26.
Muy recientemente Cool et al27 han demostrado, mediante métodos inmunohistoquímicos, la presencia del virus herpes humano tipo 8 (HHV-8) en el tejido pulmonar de 10 pacientes con HTPP. Dicho hallazgo no se observaba en los pacientes con HTP secundaria incluidos en el mismo estudio. El HHV-8 es un virus vasculotrópico implicado en el sarcoma de Kaposi. Este estudio muestra una clara asociación entre este virus y la HTPP. Estos interesantes hallazgos apuntan a que la infección por el HHV-8 en los pulmones podría ser un factor que contribuiría al crecimiento monoclonal de células endoteliales y a las mutaciones somáticas presentes en las lesiones plexiformes de los individuos con HTPP, lo que condicionaría una alteración de la función endotelial y, partiendo de este punto, todas las alteraciones que favorecen el establecimiento de la enfermedad.
Pese a los últimos hallazgos en la etiopatogenia de la HTPP, aún queda mucho por investigar en cuanto a su fisiopatología, pero cada día se avanza un poco más en la caracterización del proceso fisiopatológico de la enfermedad, lo que permite nuevas opciones terapéuticas. Efectivamente, la disfunción endotelial es, con mucho, el acontecimiento fisiopatológico central en la HTPP, ya que todos los procesos fisiopatológicos implicados en la HTPP son causa o consecuencia de ésta. Debido a su papel como órgano central de la regulación del tono vascular, todo factor que afecte al endotelio y a la liberación de los mediadores vasomoduladores que éste produce afectará también la resistencia vascular y sería causa de HTP. Los descubrimientos de los procesos genéticos, humorales y hasta víricos que pueden participar de forma importante en el establecimiento de la disfunción endotelial nos dan una idea de la complejidad de la patogenia de esta enfermedad y de la heterogeneidad de los mecanismos involucrados, condiciones que hacen de la HTPP una enfermedad incurable y de difícil tratamiento, situación que el avance de las investigaciones científicas en este campo aspira modificar.







