


Pulmonary arterial hypertension (PAH) is a heterogenous, low prevalence, severe disease.1 According to the current ESC/ERS 2022 guidelines, the disease is divided into several clinical subgroups.2 One of the subgroups is known as heritable PAH, which is caused by “de novo” or cross-generational mutation of several genes, including BMPR2, ALK, TBX4, EIF2AK4, SOX17 or SMAD9, among others. While some genes, such as those mentioned above, have demonstrated their pathogenicity, the association between many other genes and the development of PAH is under investigation.3 Although mutation of some genes may give a characteristic syndromic expression (e.g. TBX4), the clinical and/or haemodynamic features are usually not distinguishable from idiopathic PAH (iPAH), so genetic testing is recommended in iPAH, anorexigen-associated PAH, pulmonary veno-occlusive disease (PVOD) or in individuals with a prior history of familial PAH with an identified mutation.2 Importantly, previous studies have shown that patients with PAH and a positive genetic test in BMPR2 are younger, have higher mortality, greater need for lung transplantation, more severe haemodynamic parameters and lower probability of having a positive vasoreactivity test.4
The aim of this work is to determine the clinical, analytical, echocardiographic, spirometric and haemodynamic variables that characterise patients with heritable PAH and to construct a predictive model based on a nomogram to determine which subjects diagnosed with PAH are more likely to have genetic mutations.
We conducted a single-centre retrospective study in a tertiary care centre with expertise in pulmonary hypertension. Data was collected from 823 patients between 2006 and 2023 with diagnosis of PAH, of whom 302 patients were diagnosed with iPAH prior to genetic testing. We selected and analysed the data of these 302 patients. From the 302 patients diagnosed with iPAH prior to genetic testing, 59 of them had a significant genetic result (Pathogenic (P), Likely pathogenic (LP) or Variant of Unknown Significance – VUS – according to the classification of the American College of Medical Genetics and Genomics).5 All patients meet current criteria for precapillary pulmonary hypertension (mPAP>20mmHg, PVR>2 and PAWP≤15mmHg). A univariate analysis was performed with the variables collected and then a multivariate logistic regression model. Following these results, we designed a nomogram that establishes a percentage for each individual according to their characteristics that indicates the probability of the subject diagnosed with iPAH to have a genetic mutation related to this pathology. The statistical package used was Stata 14.0.
Of the fifty-nine genetic variants identified, 42 were classified as P or LP (29 in BMPR2, 4 in TBX4, 2 in ABCC8, KCNK3, and in GDF2, 1 in KCNA5 and ENG, and 1 digenic variant in KCNK3/NOTCH3 – this second is VUS). 17 patients had a VUS (4 NOTCH3, 2 in CAV1 and SMAD9, and 1 in TBX4, ABCC8, GDF2, ACVRL1, CPS1, KCNA5 and SMAD1).
Positive testing for genetic variants was associated with younger age (mean 37.7 vs 47.9 years old; p<0.001), higher haemoglobin (haemoglobin (Hb) (15.3 vs 14.2; p<0.001), higher DLCO (75% vs 66%; p=0.028), less time to first syncopal event (30.5% vs 17.8%; p=0. 03), less frequent previous or current smoking (22.5% vs 43.9%; p=0.016), hypertension (HT) (10% vs 30.5%; p=0.012) and type 2 diabetes mellitus (DM2) (0% vs 12. 5%, p=0.014). Mean pulmonary arterial pressure (mPAP) (60mmHg vs 51mmHg; p<0.001) and pulmonary vascular resistance (PVR) (13.5 WU vs 11.1 WU; p=0.008) were higher compared to patients with a negative genetic test. There were no differences in echocardiographic metrics such as tricuspid annular plane systolic excursion (TAPSE), right ventricle basal diameter, systolic pulmonary artery pressure (sPAP), TAPSE/sPAP, right atrial area and diastolic eccentricity index. In the multivariate analysis, only Hb≥15g/dL and age<40 years old were found to be statistically significant for having a genetic variant present (p=0.014 and p=0.035, respectively). DM2 was excluded from the model due to being a perfect predictor and PVR due to being a measure derived from mPAP.
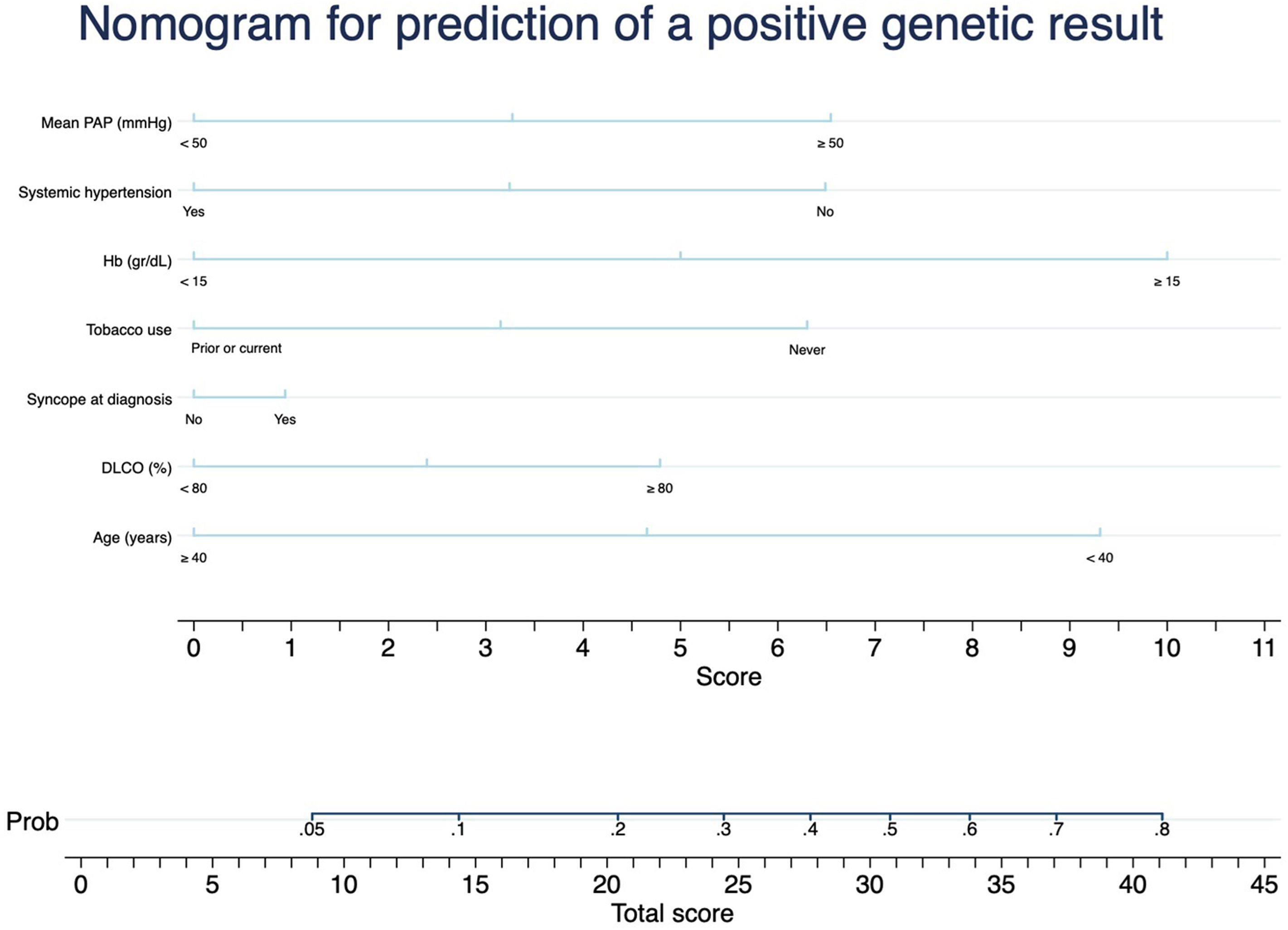
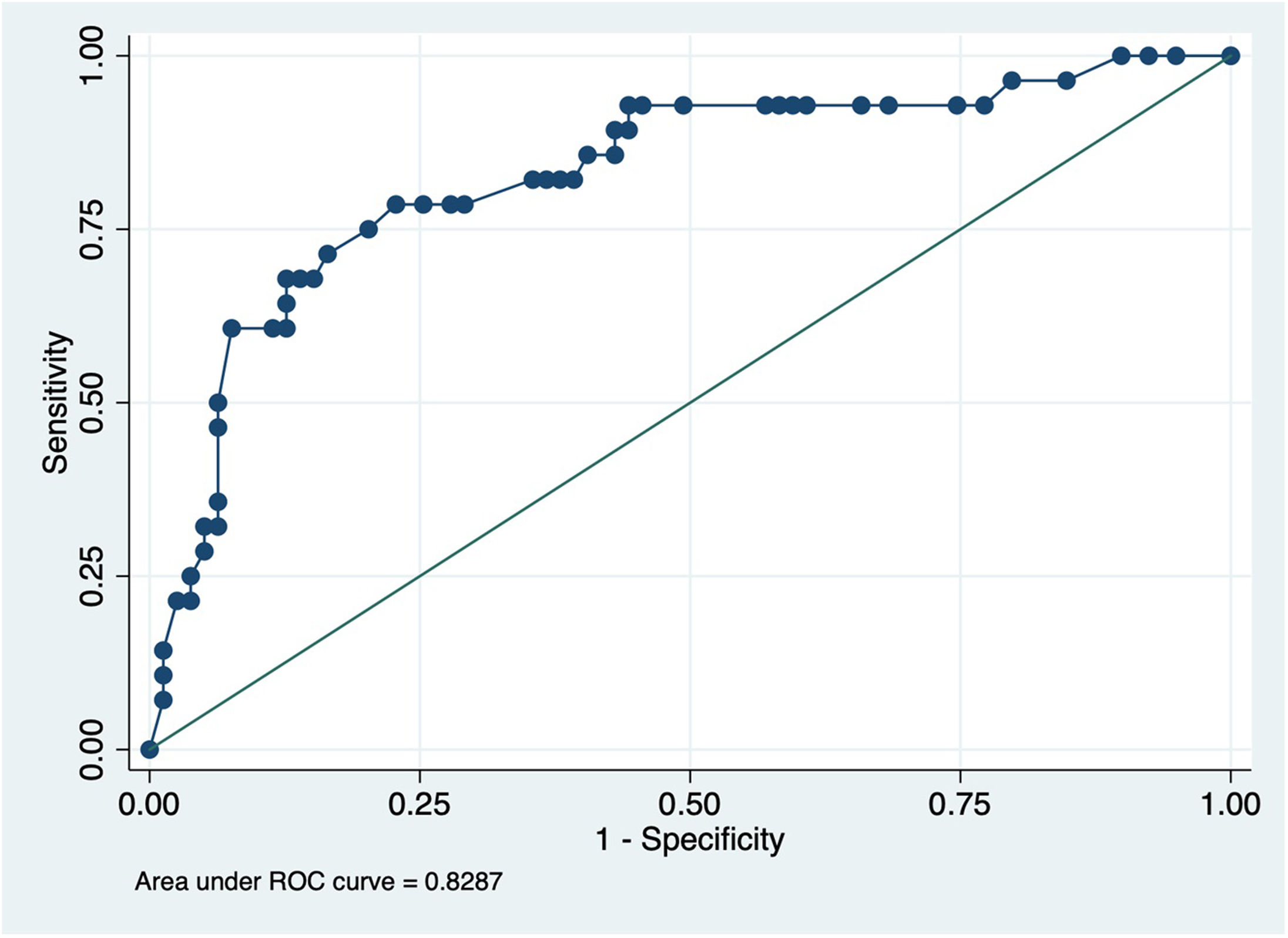
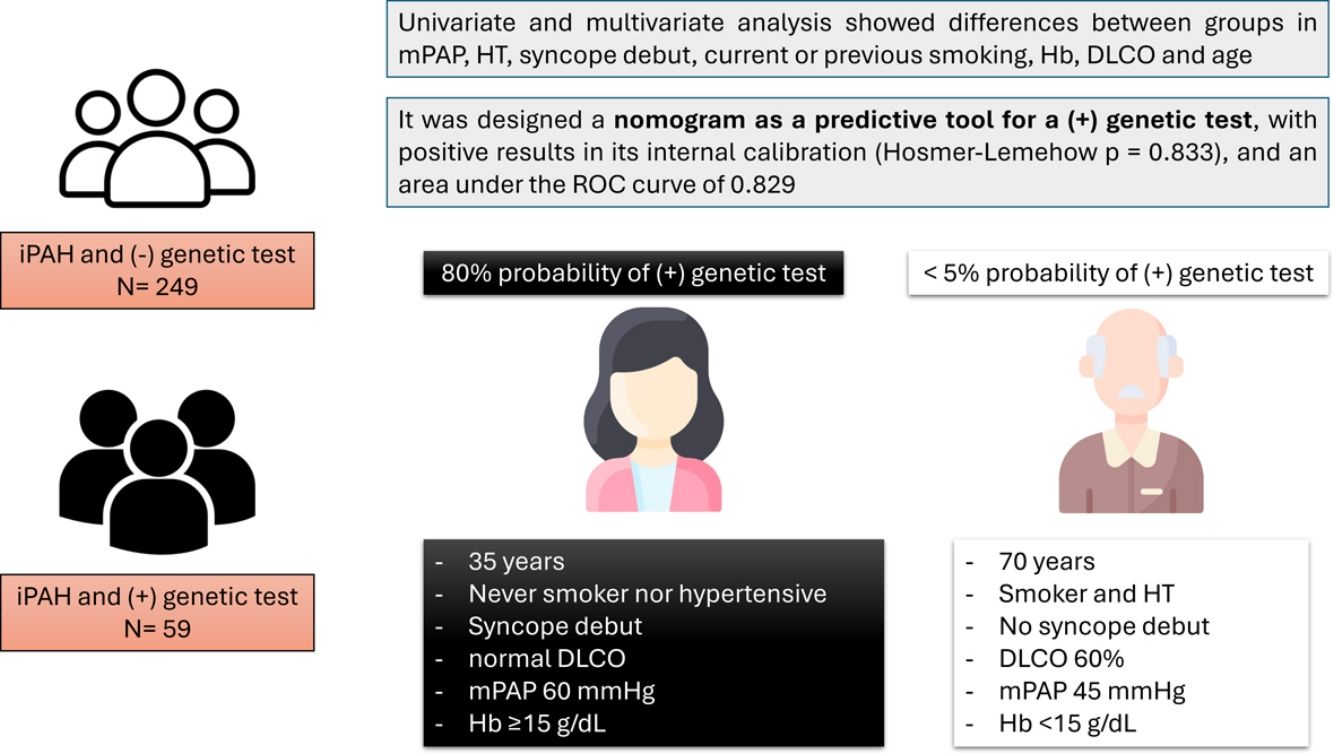
Considering the results, we designed a nomogram as a predictive tool for a positive genetic test (Fig. 1): mPAP≥50, HT, syncope debut, current or previous smoking, Hb≥15g/dL, DLCO≥80% and age≤40 years. This nomogram showed a positive result in its internal calibration (Hosmer–Lemehow goodness-of-fit test p=0.833), and an area under the ROC curve of 0.829 (95% CI 0.734–0.923) (Fig. 2).


To the authors’ knowledge, this is the first publication in the medical literature to propose a genetic mutation predictive tool for iPAH. This study compares the clinical, laboratory tests, echocardiographic, spirometric and haemodynamic characteristics of subjects with diagnosis of iPAH and a significant genetic mutation (n=59) with those without mutations at diagnosis (n=249), to find differences between the two patient profiles and thus distinguish between them prior to genetic testing.
Following the results obtained by our group, which establish mPAP≥50mmHg, age<40, Hb≥15g/dL, DLCO<80%, HT, previous or current smoking and syncope debut as variants that can help differentiate between heritable PAH and iPAH, it was decided to design a nomogram that would establish the probability of having a positive genetic test in a patient with suspected iPAH. This would allow the PH clinician to have an objective value to help him/her make the decision whether to order a genetic test for a patient or to prioritise genetic testing of one patient over another. As an example, a woman under 40 years of age, never smoker nor hypertensive, with a syncope debut, normal DLCO, mPAP 60mmHg and Hb≥15g/dL has a probability of a positive genetic test of approximately 80%, whereas a 60-year-old male, smoker, with HT, debut without syncope, DLCO 60% and Hb<15g/dL would have a probability<5%.
It is well known that heritable PAH is associated with younger patients, who have fewer comorbidities and present with greater severity.4 Therefore, it is not surprising that time to first syncopal event is sooner in younger patients with more severe disease associated with the presence of genetic variants. On the other hand, older patients without genetic variants appear to have higher rates of systemic hypertension, diabetes and lower DLCO. Additionally, we believe that haemoglobin, one of the two variables independently related with the outcome after multivariate analysis could be reflecting a greater hypoxic load during exercise in patients with genetic variants compared to patients with iPAH. The identification of Hb as a predictor of genetic background is consistent with previous studies. BMPR2,6–8TBX49 or SOX1710 genetic variants have been reported to be associated with cardiac septal defects, intrapulmonary arteriovenous malformations or parenchymal abnormalities, aspects linked with the presence of polyglobulia, and with the development of thoracic collaterals, which can increase the shunt effect, and thus the polyglobulia.
Although the evidence regarding VUS variants is scarce, family screening is still mandatory in the case of detecting a VUS. In addition, the current classification is susceptible to change with future case reports, so discarding these VUS due to lack of evidence could leave out many patients who could benefit from the proposed nomogram and would prevent the detection of a different phenotype than classic heritable PAH, if any.
In conclusion, this predictive model can identify subjects more prone to having a positive genetic test. Its application, with external validation, could be important to establish guidelines and priority on when to order genetic testing in patients with iPAH, or even modify current clinical practice, as a cut-off point could be set at which the diagnosis of iPAH can be confirmed without the need for genetic testing.
Artificial Intelligence InvolvementNo.
Funding- -
Alejandro Cruz-Utrilla holds a research contract Juan Rodes from the Instituto de Salud Carlos III, Ministerio de Ciencia, Innovación y Universidadades, Spanish Government (JR23/00071).
- -
Pilar Escribano-Subías: Grant from Instituto de Salud Carlos III PI21/01690 (Ministry of Science, Innovation &Universities, Spanish Government).
The authors declare not to have any conflicts of interest that may be considered to influence directly or indirectly the content of the manuscript.







