




Mediante el análisis de un caso de discordancia entre el nivel de alfa-1-antitripsina (AAT) en suero y el genotipo para los alelos deficientes más frecuentemente asociados al déficit de AAT (PI*S y PI*Z), se ha identificado por primera vez fuera de Portugal un paciente que presenta el alelo nulo PI*Q0ourém, el cual ha sido asociado a casos graves de enfisema pulmonar. La puesta a punto de un ensayo clínico para la detección de la mutación c.1130insT, basado en sondas fluorescentes de tipo HybProbe®, ha permitido detectar otros 4 sujetos portadores del alelo PI*Q0ourém entre un conjunto de 43 pacientes que mostraban niveles séricos de AAT anormalmente bajos atendiendo a su genotipo para los alelos PI*S y PI*Z. Puesto que 4 de los 5 casos se concentran en una misma localidad de la isla de La Palma (España), es aconsejable realizar estudios genéticos familiares y quizás un cribado poblacional localizado.
By analysis of a case of discrepancy between serum alpha-1-antitrypsin (AAT) level and genotype for the most common defective alleles associated with AAT deficiency (PI*S and PI*Z), a patient carrying the allele PI*Q0ourém has been identified for the first time outside of Portugal. This null allele has been implicated in cases of severe pulmonary emphysema. After developing a clinical assay for detection of c.1130insT mutation, based on fluorescent probes (HybProbe®), another 4 carriers of PI*Q0ourém allele were identified among 43 patients with abnormally low serum AAT levels based on their genotypes for PI*S and PI*Z alleles. Since 4 out 5 cases are from the same locality (La Palma Island, Spain), it is advisable to conduct genetic analyses of affected families and, possibly, a focused population screening.
El déficit de alfa-1-antitripsina (DAAT) es uno de los trastornos hereditarios más frecuente en las poblaciones caucasianas. Un nivel sérico de alfa-1-antitripsina (AAT) inferior a 80mg/dl, obtenido por nefelometría, incrementa el riesgo de sufrir enfisema pulmonar en la edad adulta1. Las variantes deficientes del gen SERPINA1 más comunes son los alelos PI*S y PI*Z, cuya frecuencia en la población española es del 10,4 y del 1,7%, respectivamente1. El 95% de los casos clínicos relacionados con el DAAT están asociados al genotipo PI*ZZ, y el 5% restante a los genotipos PI*SZ, PI*MZ o a combinaciones de PI*S o PI*Z con otros alelos deficientes o nulos extremadamente raros2. Estos alelos raros representan el 1,6% de las variantes deletéreas anotadas en el Registro Español de Pacientes con Déficit de AAT3. Las mutaciones nulas comprometen la estabilidad del ARN mensajero o de la proteína haciendo que la cantidad sérica de AAT sea extremadamente baja o indetectable2. Por ello, los genotipos constituidos por alelos nulos en homocigosis o acompañados de otros alelos deficientes del gen SERPINA1 conllevan un riesgo particularmente alto de padecer enfisema pulmonar de inicio muy temprano4. Presentamos aquí el caso de un paciente diagnosticado de DAAT y portador del alelo nulo PI*Q0ourém. Es la primera vez que se identifica este alelo fuera de Portugal.
Observación clínicaCaso 1Varón de 38años de edad oriundo de La Palma, sin hábitos tóxicos ni exposición laboral a irritantes respiratorios, que acude a la consulta de neumología por tos persistente tras una infección respiratoria, acompañada de sibilancias y disnea. Las pruebas iniciales practicadas, incluyendo función pulmonar, radiografía de tórax y analítica, fueron normales. Se pautó tratamiento con esteroides inhalados, pero debido a la persistencia de la sintomatología se decidió ampliar el estudio. Las pruebas adicionales incluyeron estudios inmunológicos (ANA, ENA y ANCAS), tomografía axial computarizada de alta resolución, test de la marcha y concentración sérica de AAT mediante nefelometría. Esta última prueba fue la única que mostró un resultado anormal, arrojando un valor de 38,6mg/dl (referencia: 100-180)1, motivo por el que se analizó la presencia de los alelos PI*S y PI*Z. El genotipo del paciente (PI*MS; M=no-S/no-Z) fue discordante con el nivel notablemente disminuido de AAT en suero. Ante estos resultados, se realizó el análisis molecular del gen SERPINA1 centrado en la región codificadora del alelo PI*no-S mediante PCR específica de alelo y secuenciación. Este análisis reveló la presencia del alelo PI*Q0ourém (fig. 1). Para nuestro conocimiento, es la primera vez que se describe el alelo PI*Q0ourém fuera de Portugal. Este alelo nulo, descrito por Seixas et al.5, se caracteriza por la inserción de un nucleótido de timina en la región codificadora del exón5 en el seno de un pequeño microsatélite (T)5, dentro del fondo genético normal M3. El consiguiente desplazamiento del marco de lectura genera un codón de parada prematuro que acorta la proteína 19aminoácidos en su extremo carboxilo-terminal, incluyendo un residuo de prolina esencial para la secreción de la AAT. La mutación, sin afectar al tamaño o la estabilidad del ARN mensajero, hace que la proteína alterada sea retenida en el retículo endoplásmico y degradada5.
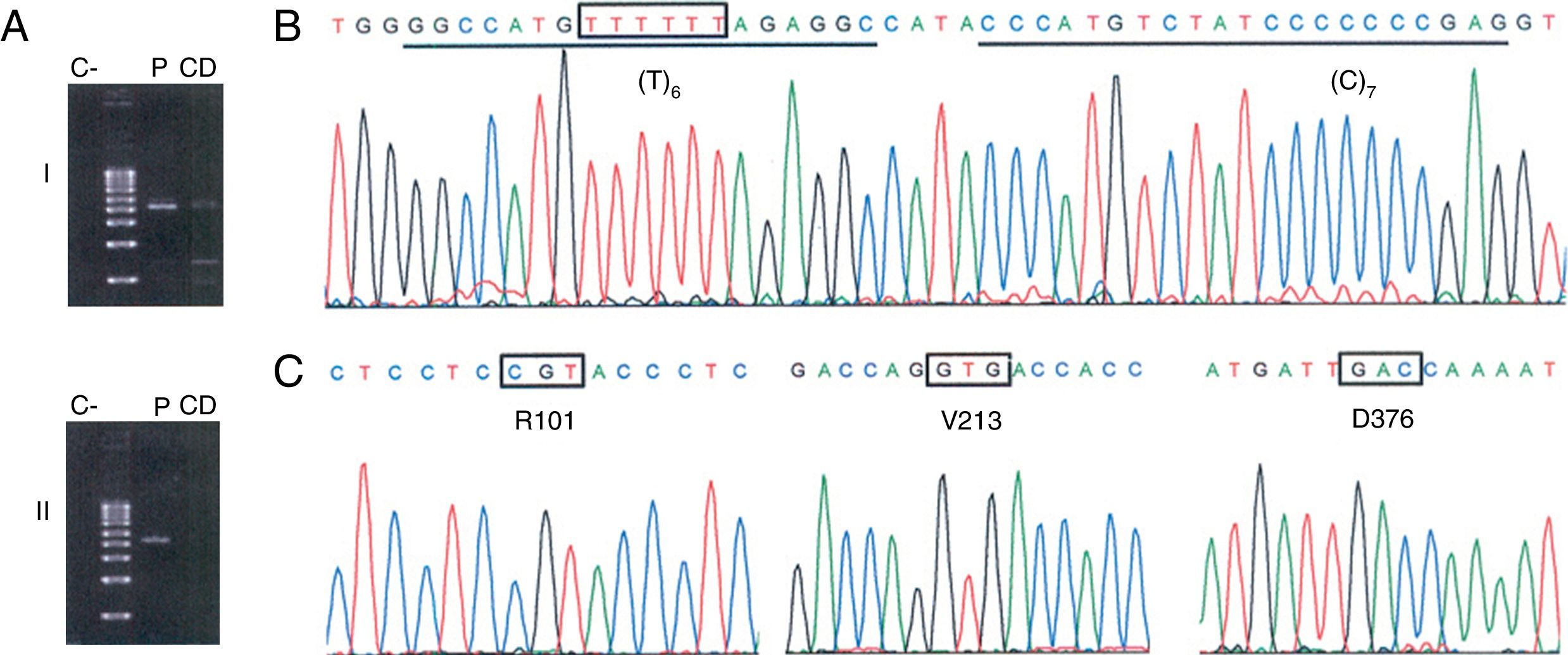
Identificación del alelo nulo PI*Q0ourém. A)Amplificación selectiva del alelo PI*no-S en 2 amplicones solapantes que cubren toda la región codificadora del gen SERPINA1 (NC_000014) y los intrones correspondientes. El fragmentoi (2.458pb; corriente arriba del sitio no-S) se amplificó con la pareja de cebadores TACTTGGCACAGGCTGGTTT//TACTTGGCACAGGCTGGTTT, y el fragmentoii (2.580pb; corriente abajo del sitio no-S) con la pareja GGGAAACTACAGCACCTGGA// GGCAGGGACCAGCTCAAC. Se resalta en negrita la posición 3’ del cebador específico de alelo que discrimina al alelo PI*S, lo cual requirió la optimización de la temperatura de anillado durante la PCR (70°C) (C: control negativo; CD: control de discriminación consistente en ADNg de un individuo con genotipo PI*SS; P: ADNg del paciente del caso 1). B-C)Electroforegramas de secuenciación del alelo PI*no-S amplificado mediante PCR específica de alelo. En B se indica con una caja el sitio afectado por la inserción de un nucleótido de timina, y se subrayan las secuencias de unión de las sondas diseñadas para la detección de dicha mutación. En C se muestran los codones y aminoácidos que definen el fondo genético M32.
Tras la puesta a punto de un ensayo para la detección de la mutación nula implicada en la generación del alelo PI*QOourém (fig. 2), este método se aplicó a un conjunto de pacientes de la consulta de neumología cuyos niveles de AAT en suero estaban por debajo del rango de referencia1 atendiendo su genotipo para los alelos PI*S y PI*Z. Este análisis se realizó sobre 43 sujetos, incluyendo 13 PI*MM, 28 PI*MS y 2 PI*MZ, y condujo a la detección de 4 nuevos casos de portadores de la mutación c.1130insT, comprobándose la presencia del fondo genético M3 mediante secuenciación. Se trata de 4 mujeres con función pulmonar y hepática normal. Sus genotipos, nivel sérico de AAT (nefelometría), edad, síntomas de enfermedad respiratoria y hábito tabáquico son los siguientes. Caso 2: Pi*MQ0ourém, 92,1mg/dl, 32años, asintomático, fumadora; caso 3: Pi*MQ0ourém, 70,5mg/dl, 58años, asintomático, no fumadora; caso 4: Pi*MQ0ourém, 65,8mg/dl, 75años, asma bronquial, no fumadora; caso 5: Pi*ZQ0ourém, 14,5mg/dl, 57años, asintomático, no fumadora.
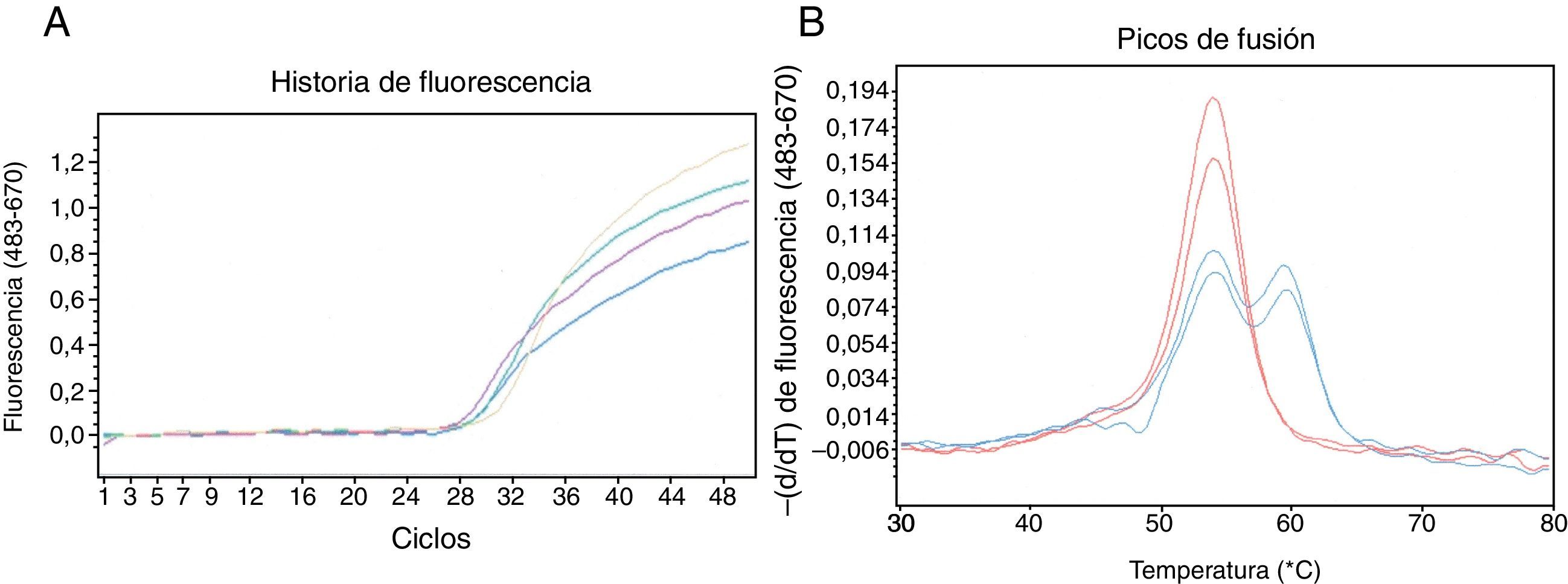
Detección de la mutación c.1130insT en el gen SERPINA1 usando sondas HybProbe®. A)Seguimiento a tiempo real del proceso de amplificación de un fragmento de ADN de 384pb. B)Resultado del análisis de fusión de los dúplex sondas/amplicón obtenidos con muestras de ADNg de 4 pacientes: 2 homocigóticos para la variante salvaje (rojo) y 2 heterocigóticos portadores de la mutación c.1130insT (azul). Los oligonucleótidos empleados fueron: CGCTTCCTGGGAGGTGT (cebador directo), TGGGAGGGATTTACAGTCACA (cebador reverso), GGCCATGTTTTTTAGAGGC (sonda sensora; fluoresceína en 3’) y CCCATGTCTATCCCCCCCGAG (sonda ancla; Cy5 en 5’ y fosfato en 3’). La mezcla de reacción (20μl) contenía la muestra de ADNg, 0,2μM de cada uno los 4 oligonucleótidos mencionados, 200μM de cada dNTP, 3mM de MgCl2, tampón de reacción 1X y 0,2μl de ADN polimerasa (Phire® II, Finnzymes). Las reacciones fueron incubadas en la plataforma LightCycler480 (Roche). Después de una desnaturalización inicial a 98°C durante 30s, se aplicaron 50ciclos de amplificación con el siguiente perfil térmico: desnaturalización, 98°C, 5s; anillado de oligonucleótidos, 50°C, 5s; extensión de cebadores, 72°C, 10s. La amplificación fue monitorizada realizando una captura de fluorescencia (excitación a 483nm y detección a 670nm) al final de cada etapa de anillado. Tras la amplificación se realizó el análisis de fusión, que consistió en: 95°C, 1min; 30°C, 1,5min; calentamiento gradual hasta 80°C realizando 3 capturas de fluorescencia por cada °C. Los datos de la curva de fusión fueron procesados con el programa de genotipado instalado en la plataforma de análisis.
Cuatro de los 5 individuos portadores del alelo PI*Q0ourém detectados en este estudio proceden de una pequeña localidad. Su presencia en la isla de La Palma probablemente está relacionada con el importante componente portugués en la colonización del archipiélago canario. Dada la gravedad de los cuadros de enfisema pulmonar asociados a genotipos homocigóticos para el alelo nulo PI*Q0ourém6, es aconsejable activar un programa preventivo que identifique a los sujetos con alto riesgo para instarles a adoptar estilos de vida saludables y ofrecerles consejo genético. En este sentido es muy útil un ensayo de detección específico para los alelos deficitarios raros más frecuentes en cada población3. El método de genotipado que se describe en este artículo es simple y de bajo coste, permitiendo una buena resolución de las variantes mutante y salvaje (aproximadamente 5,5°C). Como la sonda sensora es totalmente complementaria a la variante mutante, esta siempre originará la curva con la mayor temperatura de fusión, siendo identificada con total confidencia dentro de un alelo PI*Q0ourém o PI*Q0mattawa (fondo genético M1-Val213). Cabe destacar que la sonda ancla cubre un punto caliente de mutación (microsatélite (C)7, fig. 2), por lo que potencialmente el ensayo permitiría detectar otros alelos nulos como la PI*Q0clayton (c.1158insC en M1-Val213), PI*Q0saarbruecken (c.1158insC en M1-Ala213) o PI*Q0bolton (c.1158delC en M1-Val213). En tal caso el alelo mutante daría lugar a un pico con menor temperatura de fusión que la variante salvaje en el ensayo de genotipado.
FinanciaciónGrifols International S.A.
Conflicto de interesesLos autores declaran no tener conflictos de intereses relacionados directa o indirectamente con los contenidos del manuscrito.
Los autores agradecen a Grifols International S.A. la financiación de este trabajo de investigación.







