




La afectación del tracto respiratorio es frecuente en algunos tipos de vasculitis, fundamentalmente en las asociadas a anticuerpos anticitoplasma de neutrófilo. La presentación clínica, radiológica e histopatológica también es heterogénea y condiciona la evolución. Es necesaria, por tanto, una orientación clínica y diagnóstica precoz ya que, gracias a los nuevos tratamientos, y a pesar de ser enfermedades potencialmente graves, su pronóstico ha mejorado de manera considerable en los últimos años.
En el presente trabajo se realiza una actualización del diagnóstico y las nuevas opciones terapéuticas de las vasculitis pulmonares.
Respiratory tract affectation is frequent in some types of vasculitis, fundamentally in those associated with anti-neutrophil cytoplasmic antibodies. The clinical, radiological and histopathological presentation is also heterogeneous and conditions the evolution. It is therefore necessary to establish an early diagnosis based on the symptoms because, thanks to new treatments, and despite them being potentially serious diseases, their prognosis has improved considerably in recent years.
The present paper updates the diagnosis and the new therapeutic options for pulmonary vasculitis.
Las vasculitis sistémicas son un grupo muy heterogéneo de enfermedades que se caracterizan por una inflamación de la pared de los vasos. Se ha descrito la presencia de un infiltrado inflamatorio, agudo o crónico, que conduce a la desestructuración de los vasos sanguíneos con disminución del flujo sanguíneo a los tejidos1,2. Pueden afectar a uno o a varios órganos o sistemas dependiendo del tamaño y de la localización de los vasos afectados, y la presentación clínica, por tanto, puede ser proteiforme.
Históricamente las vasculitis se han clasificado en función de su histopatología (figs. 1 y 2)3. En la conferencia de Chapel Hill4 se propuso una nomenclatura y una definición para las vasculitis sistémicas (tabla 1). Esta clasificación se basa en el tamaño de los vasos implicados e incorpora la existencia de los anticuerpos anticitoplasma de neutrófilo (ANCA). La vasculitis de células gigantes, poliarteritis nudosa y enfermedad de Kawasaki incluidas en dicha clasificación, raramente presentan afectación pulmonar4,5. Por el contrario, otras vasculitis no incluidas en la clasificación de Chapel Hill, pero que sí tienen afectación pulmonar, son las asociadas a enfermedades autoinmunes sistémicas, como la enfermedad de Behçet o el lupus eritematoso sistémico. El consenso de Chapel Hill aún está vigente, ya que ofrece un marco útil para clasificar las vasculitis según el tamaño del vaso dañado, que permite una aproximación a las diferentes presentaciones clinicopatológicas.
.")
Clasificación de las vasculitis. ANCA: anticuerpos anticitoplasma de neutrófilo; IF: inmunofluorescencia. (Tomada de Gómez-Román3).
. ANCA: anticuerpos anticitoplasma de neutrófilo; IF: inmunofluorescencia. (Tomada de Gómez-Román5).")
Clasificación de las vasculitis (continuación). ANCA: anticuerpos anticitoplasma de neutrófilo; IF: inmunofluorescencia. (Tomada de Gómez-Román5).
Clasificación de las vasculitis sistémicas según el consenso de Chapel Hill
| Vasculitis de grandes vasos |
| Arteritis de células gigantes |
| Arteritis de Takayasu |
| Vasculitis de mediano vaso |
| Poliarteritis nodosa |
| Enfermedad de Kawasaki |
| Vasculitis de pequeño vaso |
| Granulomatosis de Wegenera |
| Síndrome de Churg-Straussa |
| Poliangeítis microscópicaa |
| Púrpura de Schönlein-Henoch |
| Crioglobulinemia |
| Angeitis cutánea leucocitoclástica |
El aparato respiratorio puede verse afectado en las vasculitis sistémicas, aunque de manera más frecuente en las vasculitis asociadas a ANCA, como veremos a continuación.
Formas de afectación pulmonar en las vasculitisLas vasculitis pulmonares comprenden un grupo heterogéneo de enfermedades muy diferentes en su presentación tanto clínica como radiológica, y que se asocian frecuentemente con la presencia de afectación sistémica6. A pesar de incluir entidades tan diferentes, existen síntomas y signos comunes que son sugestivos y que, junto con los datos biológicos, permiten aventurar un diagnóstico diferencial más específico7–9.
Pueden afectar a los pequeños vasos del pulmón (capilaritis pulmonar) y ocasionar una hemorragia alveolar difusa, caracterizada por la triada de infiltrados alveolares difusos, hemoptisis y caída en el hematocrito y/o hemoglobina10. También se ha asociado con algunos tipos de vasculitis pulmonares (vasculitis ANCA) las presencia en las pruebas de imagen de cavidades o nódulos, así como inflamación de los grandes vasos pulmonares que conlleva la aparición de aneurismas y trombosis11.
Hemorragia alveolar difusaLa afectación pulmonar de las vasculitis puede desarrollarse en cualquier vaso, independientemente del tamaño. La vasculitis de la microvasculatura pulmonar se conoce como capilaritis pulmonar. Aunque se trata de un diagnóstico patológico, su presencia hace necesario descartar algún tipo de enfermedad subyacente12, ya que puede ser la primera manifestación, y es frecuente encontrarla en las vasculitis que presentan ANCA o enfermedades autoinmunes sistémicas. La complicación más grave de la capilaritis pulmonar, consecuencia del daño en la microcirculación, es la hemorragia pulmonar. Aunque la hemorragia alveolar pulmonar puede deberse a un daño alveolar difuso por múltiples etiologías, en el caso de las vasculitis es consecuencia de la inflamación de los propios capilares pulmonares13.
Se caracteriza por la presencia de un infiltrado intersticial con neutrófilos y necrosis fibrinoide en la pared de los capilares14. Secundariamente a esa inflamación y a la presencia de neutrófilos, se produce una pérdida de integridad en la membrana basal de la unión alveolocapilar con disrupción de la misma y extravasación de hematíes a los espacios alveolares. Es muy típica la presencia de hemosiderófagos en el lavado broncoalveolar, ya que los macrófagos alveolares fagocitan los eritrocitos que fluyen a través de la lesión en la membrana basal del endotelio vascular hasta el alvéolo15.
La hemorragia pulmonar se ha descrito como la manifestación más grave de las vasculitis asociadas a ANCA y es más frecuente en la poliangeítis microscópica (PAM)16,8 y en la granulomatosis de Wegener17–20. También se ha asociado, aunque con menor frecuencia, al síndrome de Goodpasture21, al síndrome de Churg-Strauss22–25, a la púrpura de Schönlein-Henoch26–28, a la arteritis de Takayasu, a la arteritis de células gigantes, a la crioglobulinemia, a la poliarteritis nudosa o a la enfermedad de Behçet29,30 (tabla 2)31. Desde el punto de vista clínico se define como un síndrome caracterizado por la presencia de hemoptisis —aunque puede no estar presente en un tercio de los casos32—, anemia, insuficiencia respiratoria aguda e infiltrados alveolares pulmonares de aparición brusca33. Es una complicación potencialmente mortal y con una evolución clínica impredecible. Asimismo, pueden aparecer otros síntomas más inespecíficos, como dolor torácico, tos, fiebre o síndrome general semanas o meses antes.
Afectación pulmonar y vasculitis
| Vasculitis con afectación pulmonar frecuente | Enfermedad de Churg-StraussEnfermedad de WegenerPoliangeítis microscópica |
| Vasculitis con afectación pulmonar infrecuente | Púrpura de Schönlein-HenochArteritis de TakayasuArteritis de células gigantesCrioglobulinemiaPoliarteritis nudosaEnfermedad de Behçet |
Adaptado de Fishbein31.
La radiografía de tórax es inespecífica, muestra infiltrados alveolares localizados o difusos y en muchas ocasiones requiere la realización de tomografía computarizada de tórax de alta resolución para definir y confirmar los hallazgos. Las pruebas de función respiratoria se caracterizan por un aumento en la capacidad de difusión del monóxido de carbono (DLCO), aunque en ocasiones no es factible si el paciente se encuentra en situación crítica34. La realización de una fibrobroncoscopia con lavado broncoalveolar es obligada, si la situación clínica del enfermo lo permite, ya que ayuda a establecer el diagnóstico y a descartar otras entidades. La presencia de más de un 20% de hemosiderófagos en el lavado broncoalveolar es diagnóstica de hemorragia alveolar pulmonar incluso en los casos con presentación subclínica.
El abordaje terapéutico de los pacientes con hemorragia alveolar pulmonar dependerá de la gravedad de la misma, así como de las manifestaciones extrapulmonares del cuadro de vasculitis. En los casos de gravedad puede ser necesario el traslado del paciente a una unidad de cuidados intensivos con soporte respiratorio.
Se han propuesto diferentes sistemas de clasificación en función de la gravedad, y de esta depende la intensidad del tratamiento. En todos los casos se realiza un tratamiento de inducción que trata de controlar la actividad de la vasculitis y alcanzar un estado de remisión en unas semanas, seguido de una fase de mantenimiento para prevenir las recidivas35.
El tratamiento de inducción en los casos en que el paciente presenta estabilidad y no precisa soporte ventilatorio se basa en regímenes de terapia combinada que incluyen inmunosupresores como glucocorticoides y ciclofosfamida, metotrexato36 o rituximab37,38. Estos dos últimos fármacos han demostrado mayor eficacia y menos efectos secundarios que la primera, aunque es necesario más tiempo de tratamiento para lograr la remisión. El rituximab se está planteando recientemente como una alternativa a la ciclofosfamida, ya que se han demostrado tasas de eficacia similares con los dos inmunosupresores39. En los casos con afectación más grave, además del tratamiento anteriormente descrito, se ha planteado la plasmaféresis como opción terapéutica. Sin embargo, no existe evidencia científica sólida que la apoye. Para los casos refractarios al tratamiento previo se ha ensayado el uso de inmunoglobulinas y de rituximab, con buenos resultados40,41.
En el tratamiento de mantenimiento se han empleado glucocorticoides a dosis bajas, ya que se ha demostrado que previenen las recidivas42. Asociados a estos, para poder mantener dosis bajas, tras 3 a 6 meses de tratamiento con ciclofosfamida puede sustituirse esta por otro inmunosupresor, como el metotrexato o la azatioprina43. Otros tratamientos, como el micofenolato de mofetil o la leflunomida, han demostrado ser menos eficaces que la azatioprina para prevenir las recaídas44.
Enfermedad granulomatosa pulmonarLas vasculitis que producen enfermedad granulomatosa pulmonar se caracterizan por la presencia de anticuerpos tipo ANCA. Producen una inflamación granulomatosa de las vías respiratorias, altas y bajas, con presencia de vasculitis necrosante y afectación renal con hematuria y proteinuria. En la mayoría de los casos los anticuerpos van dirigidos contra la proteinasa 3 citoplasmática (c-ANCA)45. En la patogenia de este tipo de afectación pulmonar se ha implicado una respuesta celular inflamatoria que deriva en la formación de granulomas formados por un infiltrado inflamatorio compuesto por neutrófilos, linfocitos, células plasmáticas, histiocitos y eosinófilos18. Asimismo, la necrosis del propio parénquima pulmonar puede derivar en la formación de microabscesos o zonas con un centro necrótico rodeado de un anillo de histiocitos que ofrece una imagen radiológica típica (signo del halo). También se han descrito aumento de la trama broncovascular en el parénquima, adenopatías hiliares y derrame pleural asociado46. Cuando este tipo de lesiones aparecen en relación con una fase de actividad de la enfermedad se trata de lesiones inflamatorias activas que responden adecuadamente al tratamiento inmunosupresor47. Con dicho tratamiento los nódulos cavitados y los mayores de 3cm, así como los infiltrados, tienden a desaparecer y dejan pequeñas líneas de fibrosis cicatricial48. El tratamiento en este tipo de afectación es similar al descrito previamente para la capilaritis con hemorragia alveolar.
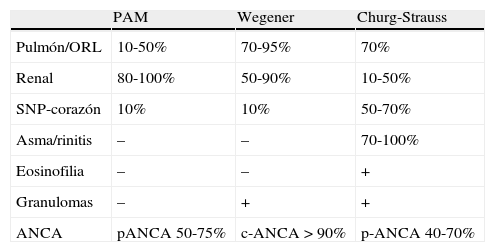
Vasculitis con afectación pulmonarEl pulmón es un posible órgano diana de las vasculitis sistémicas, y algunas de ellas —como las asociadas a ANCA— son de muy frecuente afectación (tabla 3). El término «vasculitis pulmonares» no se refiere a una única entidad, sino al proceso clínico-patológico que tiene lugar en la génesis de las manifestaciones pulmonares de estas enfermedades. En España se ha descrito la incidencia a 5 años para algunas de estas vasculitis: 2,95 casos/millón habitantes para la enfermedad de Wegener, 7,91 casos/millón habitantes para la PAM y 1,31 casos/millón habitantes para la enfermedad de Churg-Strauss49.
Características de las vasculitis asociadas a anticuerpos anticitoplasma de los neutrófilos
| PAM | Wegener | Churg-Strauss | |
| Pulmón/ORL | 10-50% | 70-95% | 70% |
| Renal | 80-100% | 50-90% | 10-50% |
| SNP-corazón | 10% | 10% | 50-70% |
| Asma/rinitis | – | – | 70-100% |
| Eosinofilia | – | – | + |
| Granulomas | – | + | + |
| ANCA | pANCA 50-75% | c-ANCA > 90% | p-ANCA 40-70% |
PAM: poliangeítis microscópica; ORL: afectación otorrinolaringológica; SNP: sistema nervioso periférico; ANCA: anticuerpos anticitoplasma de neutrófilo.
La granulomatosis de Wegener, actualmente conocida como granulomatosis con poliangeítis, es una vasculitis sistémica caracterizada por inflamación granulomatosa de la vía aérea con vasculitis necrosante en los vasos de pequeño calibre. En esta enfermedad pueden coexistir otro tipo de afectaciones, al igual que en otras vasculitis, como la renal o la nerviosa. Es muy típica la presencia de glomerulonefritis necrosante4.
La afectación del tracto respiratorio es bastante frecuente, tanto de la vía aérea superior como de la inferior50,51. En la vía aérea superior es frecuente la presencia de sensación de obstrucción nasal con rinorrea y úlceras, que en algunos casos provocan la perforación del tabique nasal. Las manifestaciones clínicas por afectación más baja son tos, disnea y/o hemoptisis con dolor torácico de características pleuríticas. Se han descrito estenosis traqueobronquiales que pueden condicionar la presencia de disnea y estridor en algunos pacientes52. Puede existir también derrame pleural, habitualmente unilateral53. Una complicación frecuente de la enfermedad de Wegener es la hemorragia alveolar pulmonar por capilaritis —que ya se ha descrito previamente— en un alto porcentaje de pacientes32. La presentación clínica puede ser subaguda o aguda, con disnea, hemoptisis y la presencia en la radiografía de tórax de infiltrados alveolares bilaterales de reciente aparición, a la vez que se detecta una caída en el hematocrito. Como se ha señalado, es característico un aumento en la capacidad de difusión de CO, así como la presencia de siderófagos en el lavado broncoalveolar54. Es muy típica, en esta vasculitis, la presencia de nódulos pulmonares múltiples, en ocasiones cavitados55, así como signos de obstrucción bronquial, infiltrados pulmonares o tractos fibróticos parenquimatosos. Los nódulos pulmonares en ocasiones presentan el signo del halo con un anillo de opacidad en vidrio deslustrado que los rodea56 y que es característico. La radiología puede ayudar a orientar si la enfermedad se encuentra en una fase activa. La presencia de patrón en vidrio deslustrado junto con nódulos pulmonares mayores de 3cm es indicativa de actividad de la enfermedad57. El diagnóstico no siempre es sencillo, ya que aunque los hallazgos radiológicos son evidentes, la rentabilidad de la biopsia transbronquial58 o de la citología del lavado bronocalveolar es baja. El mejor método para llegar al diagnóstico es la biopsia abierta. Ante la presencia de infiltrados pulmonares o nódulos es posible encontrar granulomas necrosantes y vasculitis hasta en el 90% de los casos59. Aunque muchas de las lesiones parenquimatosas mejoran tras el tratamiento, es posible encontrar lesiones cicatriciales residuales en la mayoría de los pacientes.
La enfermedad de Wegener se caracteriza por asociarse con la presencia de ANCA, que a su vez se utilizan para monitorizar la respuesta al tratamiento60. Se han descrito títulos elevados de c-ANCA en más del 90% de los pacientes con enfermedad activa generalizada, y del 40-70% de los pacientes con enfermedad activa localizada60,61. Más del 90% de los pacientes con enfermedad de Wegener presentan ANCA contra la proteinasa 3, y el 10% restante contra la mieloperoxidasa62. El cambio en los títulos de estos anticuerpos se correlaciona con la actividad de la enfermedad. Sin embargo, pueden persistir elevados en el 40% de los pacientes tras alcanzar la remisión clínica.
Poliangeítis microscópicaLa PAM es una vasculitis de pequeño vaso no granulomatosa asociada a la presencia de ANCA. Es muy frecuente la afectación renal en forma de glomerulonefritis necrosante, así como la afectación respiratoria, de presentación ya sea simultánea o aislada63. Forma parte de los síndromes renopulmonares, al igual que la granulomatosis de Wegener, y puede iniciarse así o presentarla a lo largo de su evolución64. La forma de presentación pulmonar más frecuente es la hemorragia alveolar por capilaritis, con una prevalencia en torno al 15-30%65, dependiendo de las series. La evolución de la hemorragia alveolar difusa es rápidamente progresiva en la mayoría de los pacientes, aunque una minoría pueda tener un comienzo más insidioso. Los síntomas pueden preceder al diagnóstico en uno de cada 4 pacientes63. La PAM puede afectar simultáneamente a otros órganos o sistemas, como el sistema nervioso o el musculoesquelético, pero también al tracto gastrointestinal o al corazón. Las decisiones terapéuticas deben tomarse en función de la gravedad y del patrón de afectación orgánica.
Enfermedad de Churg-StraussLa enfermedad de Churg-Strauss, también conocida como granulomatosis alérgica con angeítis, es un síndrome caracterizado por una inflamación granulomatosa con abundantes eosinófilos y vasculitis necrosante que afecta a vasos de pequeño y mediano calibre, fundamentalmente del pulmón66, y que se asocia con asma y eosinofilia19.
El curso evolutivo de la enfermedad suele presentar varias fases. En un primer momento es típica la presencia de asma alérgica con rinitis y, en ocasiones, sinusitis y poliposis nasal. Posteriormente aparece eosinofilia en sangre periférica, para finalmente presentar manifestaciones de vasculitis sistémica. La presencia de asma es casi constante, y está presente en el 95% de los pacientes67. Al ser una enfermedad que ocurre en distintas fases, el asma puede preceder a la vasculitis en varios años68. En la mayoría de los casos el asma remite tras hacerlo la vasculitis pero puede persistir años después, precisando de tratamiento corticoideo durante largas temporadas o de otros fármacos añadidos para lograr disminuir la dosis de corticoides69,70. Otras manifestaciones pulmonares menos frecuentes son la hemorragia alveolar difusa71 y la presencia de derrame pleural72. Asimismo, puede afectar a otros órganos diferentes del pulmón, como los sistemas nervioso y cardiovascular, la piel73 o el sistema musculocutáneo en forma de polimiositis74.
En la etiopatogenia de la enfermedad antiguamente se había implicado la presencia de inmunocomplejos en las paredes vasculares, pero más recientemente se ha visto que subyace una vasculitis mediada por ANCA75.
La presentación histopatológica en el pulmón puede combinar la presencia de granulomas extravasculares, vasculitis y neumonía eosinofílica76, aunque la coexistencia de las 3 lesiones es infrecuente. Los granulomas están formados con un centro de eosinófilos rodeados de un infiltrado de histiocitos y la vasculitis se caracteriza también por un infiltrado de eosinófilos en la íntima y la media de los vasos.
La radiografía de tórax muestra la presencia de infiltrados pulmonares evanescentes y parcheados sin una clara distribución lobar, así como nódulos pulmonares, raramente cavitados, u otras alteraciones como engrosamiento septal, patrón en vidrio deslustrado o derrame pleural77.
Los datos de laboratorio apoyan el diagnóstico clínico con la presencia de leucocitosis con marcada eosinofilia, aumento de reactantes de fase aguda y de IgE, que frecuentemente se correlaciona con la actividad de la enfermedad68. En la enfermedad de Churg-Strauss los ANCA son positivos en el 40-60% de los casos78. El patrón suele ser p-ANCA, al contrario que en la granulomatosis de Wegener. Con el tratamiento se observa un aumento en la supervivencia de estos pacientes79.
Otras vasculitisPoliarteritis nudosaLa poliarteritis nudosa (PAN) clásica es una vasculitis de arterias de mediano calibre, arteriolas, capilares o vénulas. Es poco frecuente la afectación del sistema respiratorio80. Se ha descrito la presencia de hemorragia alveolar pulmonar en algún caso de PAN asociada a virus de la hepatitis B, así como de infiltrados intersticiales difusos81–83. También se ha documentado algún caso con nódulos pulmonares no asociados a ANCA84. Las arterias pulmonares se afectan raramente, al contrario que las bronquiales. En estudios previos sobre la afectación pulmonar de esta vasculitis81 se describió la lesión de las arterias bronquiales con daño alveolar junto con datos de fibrosis.
CrioglobulinemiaLa crioglobulinemia es una vasculitis de pequeño vaso que se asocia a la presencia en suero de crioglobulinas y en muchos casos a virus de la hepatitis C. La afectación más frecuente es la cutánea, seguida de la renal, en forma de glomerulonefritis4. La afectación pulmonar, cuando está presente, suele ser leve. Los pacientes presentan tos, disnea de esfuerzo o dolor torácico pleurítico85. Aunque menos habitual, se ha descrito el desarrollo de hemorragia alveolar pulmonar, consecuencia de capilaritis pulmonar86–88.
En las pruebas de imagen es posible no encontrar hallazgos o, por el contrario, mostrar datos de fibrosis intersticial89, derrame pleural o infiltrados pulmonares90. Todos estos hallazgos se traducen en las pruebas de función respiratoria en una afectación restrictiva con disminución en la difusión de CO91. El lavado broncoalveolar de estos pacientes muestra un aumento de linfocitos T, consecuencia de la alveolitis subyacente.
Schönlein-HenochLa enfermedad de Schönlein-Henoch es una vasculitis de pequeño vaso debida al depósito de inmunocomplejos IgA. Es una enfermedad típica de la infancia, aunque se han descrito casos en adultos92. Las afectaciones más frecuentes son la cutánea, la renal y la intestinal. La afectación pulmonar es muy poco frecuente, aunque se han descrito casos de neumonía intersticial usual, así como casos de hemorragia alveolar pulmonar más graves93,94. Como en otras vasculitis, en presencia de daño alveolar se han descrito alteraciones de la DLCO, aumentada en el caso de la hemorragia alveolar. En la histopatología se han reportado, además de necrosis de las paredes de los capilares pulmonares, depósitos de IgA en los alvéolos próximos a las zonas de capilaritis92.
Arteritis de células gigantesLa arteritis de células gigantes es una vasculitis granulomatosa que afecta típicamente a vasos de gran calibre, como la aorta y sus ramas. Su presentación es característica de la edad avanzada, y es frecuente que se asocie con la presencia de polimialgia reumática.
La afectación pulmonar es infrecuente95. Puede presentar además síntomas generales como fiebre y astenia, tos seca96, dolor torácico pleurítico, disnea97 y, en ocasiones, derrame pleural o enfermedad intersticial pulmonar98,99. Radiológicamente la presentación es variada. Puede oscilar entre infiltrados intersticiales, nódulos pulmonares o, en los casos más graves, infiltrados alveolares debidos a hemorragia pulmonar100–102. En cualquier caso, la afectación más característica de esta vasculitis son los aneurismas vasculares, que pueden detectarse mediante pruebas de imagen y que en caso de disección conllevan una alta mortalidad103. La histopatología de esta enfermedad muestra datos de vasculitis con inflamación en la adventicia y la media de grandes vasos con presencia de células gigantes, que condiciona una necrosis fibrinoide con destrucción de la pared vascular104.
TakayasuLa arteritis de Takayasu es una vasculitis granulomatosa que afecta a vasos de gran calibre, como la de células gigantes. Al contrario que esta, afecta predominantemente a personas jóvenes. La exploración física en esta enfermedad es crucial, ya que puede sospecharse en un paciente joven con hipertensión arterial y asimetría en la exploración de los pulsos periféricos105.
La afectación pulmonar suele ser subclínica, aunque en los casos con síntomas es típica la tos o la disnea de esfuerzo como manifestaciones iniciales106. Son pocos los casos en que se ha descrito la presencia de hemorragia pulmonar por capilaritis, hipertensión pulmonar o rotura de microaneurismas107–109. Se han descrito otras manifestaciones pulmonares menos frecuentes, como derrame pleural, infiltrados pulmonares o hemorragia pulmonar110.
La histopatología muestra un infiltrado inflamatorio compuesto por células mononucleares y células gigantes que generan granulomas y condicionan la fragmentación de la media, elástica y, por tanto, de la pared vascular. Posteriormente pueden aparecer fenómenos trombóticos con obliteración de la luz del vaso y fibrosis111. Al igual que en otras vasculitis —como la de células gigantes—, la resonancia magnética112 y la tomografía con emisión de positrones (PET) han demostrado gran utilidad para el diagnóstico, así como para monitorizar la actividad de la enfermedad. En concreto, la PET es capaz de demostrar el grado de inflamación y la extensión de la misma con una alta sensibilidad113.
TratamientoEl tratamiento de las vasculitis con afectación pulmonar habitualmente incluye el uso de corticoesteroides y otros agentes inmunosupresores. Hay que tener en cuenta que cualquier tratamiento de este tipo tiene potenciales efectos adversos, por lo que deben utilizarse en función del grado de afectación de la enfermedad y así evitar posibles toxicidades.
Para clasificar el grado de actividad de estas enfermedades se han propuesto diversos sistemas. El Grupo Europeo de Estudio de las Vasculitis (EUVAS) ha propuesto una clasificación basada en 5 niveles: enfermedad localizada, enfermedad precoz generalizada, enfermedad generalizada, enfermedad grave y enfermedad refractaria. Lógicamente, se emplearán tratamientos más agresivos cuanto mayor sea el grado de actividad o de gravedad de la enfermedad. Con la introducción del tratamiento inmunosupresor la mortalidad a 5 años de los pacientes con vasculitis ha disminuido del 50 al 12% desde los años setenta50.
Tratamiento de inducciónComo tratamiento de inducción de primera línea habitualmente se utilizan los corticoides asociados a otro inmunosupresor, casi siempre ciclofosfamida. Este fármaco se utiliza en dosis de 2-3mg/kg/día114,115, que puede ajustarse en función de la aparición de los efectos adversos o de la respuesta del paciente. En las últimas décadas diversos estudios han demostrado un beneficio en la aplicación de este fármaco en pulsos cada 3-4 semanas116.
Como alternativa a la ciclofosfamida se han ensayado otros fármacos, como el metotrexato, que también ha demostrado ser un buen tratamiento inductor aunque puede no estar indicado en pacientes con afectación renal por la vasculitis. El metotrexato ha demostrado ser tan efectivo como la ciclofosfamida para la inducción, pero a su vez se han descrito mayor número de recaídas tras un año de tratamiento36.
Tratamiento de mantenimientoUna vez conseguida la remisión, existen varias opciones para el tratamiento de mantenimiento. La azatioprina y el metotrexato han demostrado ser equivalentes117 tanto en eficacia como en las tasas de recidiva de la enfermedad. Otra opción terapéutica es el micofenolato de mofetilo, que aunque ha demostrado ser un buen fármaco inductor, en un estudio en el que se lo comparó con la azatioprina no demostró superioridad respecto a ésta118, y por el contrario sí presentó mayores tasas de recidiva que la azatioprina.
PlasmaféresisLa plasmaféresis se introdujo por primera vez en los años setenta para eliminar los inmunocomplejos circulantes en la enfermedad por anticuerpos antimembrana basal glomerular119.
El planteamiento de esta técnica se basa en el descubrimiento de anticuerpos circulantes (ANCA) asociados a vasculitis que producen daño renal y pulmonar. Además de la retirada de la circulación sistémica de dichos anticuerpos, se han visto otros aspectos beneficiosos de esta técnica, como retirada de otros factores como complemento, fibrinógeno, inmunoglobulinas, moléculas de adhesión y capacidad de reposición de factores de coagulación. Además, se ha visto que puede mejorar la función renal de estos pacientes evitando su paso a diálisis.
Se han realizado estudios en los diferentes tipos de vasculitis. En pacientes con granulomatosis de Wegener se ha demostrado mejor función renal en los sometidos a plasmaféresis120, así como un aumento en la supervivencia asociada a causa renal. En pacientes con PAM existen pocos datos, aunque algunos trabajos recientes han mostrado una inducción a la remisión más rápida en pacientes con plasmaféresis asociada a inmunosupresión121.
Donde existe más evidencia sobre el uso de plasmaféresis es en los casos de hemorragia alveolar pulmonar, aunque no existen ensayos clínicos controlados. Se han reportado algunos datos contradictorios. Algunos trabajos mostraron buenos resultados, con resolución de la hemorragia alveolar difusa en el 100% de los pacientes, aunque un tercio de ellos no presentaban afectación renal por la vasculitis122. En otro trabajo, en cambio, el tratamiento con plasmaféresis se asoció con un 50% de mortalidad tras 2 años123.
Con todo esto, la plasmaféresis podría ser un tratamiento adyuvante a la inmunosupresión durante la fase aguda en las vasculitis con afectación moderada-grave, así como en los casos de enfermedad refractaria. Se recomienda la combinación de plasmaféresis, corticoides y ciclofosfamida en el tratamiento de enfermedad con afectación grave tras los datos aportados por el estudio MEPEX124, desarrollado en pacientes con vasculitis ANCA y afectación renal. Aquellos en los que se realizó plasmaféresis (69%) presentaron menor tasa de mortalidad y menor necesidad de diálisis.
Fármacos antifactor de necrosis tumoralEl factor de necrosis tumoral alfa (TNF-α) se encuentra sobreexpresado en las vasculitis. Debido a esto, se ha ensayado el tratamiento con fármacos biológicos frente a este receptor. El infliximab, aprobado para el tratamiento de otras enfermedades autoinmunes, ha demostrado buenos resultados en la inducción a la remisión en pacientes con enfermedad refractaria125,126. Sin embargo, no se ha visto que sea un tratamiento eficaz para el mantenimiento a medio o largo plazo127.
Son pocos los trabajos realizados sobre él, y se precisa una mayor experiencia clínica. En algún estudio con adalimumab únicamente se han visto requerimientos menores de corticoides en el tratamiento conjunto con ciclofosfamida en pacientes con vasculitis ANCA y afectación grace128. Con este grupo de tratamientos, paradójicamente, hace poco se ha descrito la aparición de algunos casos de vasculitis secundaria a fármacos129.
Inmunoglobulinas intravenosasSe han empleado inmunoglobulinas intravenosas como tratamiento, único o tras el tratamiento estándar previo, para los casos de vasculitis con enfermedad persistente, con el fin de mantener la remisión. El efecto de las inmunoglobulinas intravenosas es limitado en el tiempo y podría ser útil cuando existe contraindicación al resto de terapias130. Hay trabajos con buenos resultados en vasculitis ANCA, con remisiones en torno al 50% de la enfermedad en combinación con corticoesteroides y/o otros inmunosupresores131,132. Estudios más recientes ofrecen buenos datos, con tasas de remisión del 60% en la administración mensual de inmunoglobulinas junto con terapia estándar en vasculitis con enfermedad recidivante133. A la vista de estos resultados, y dada la buena tolerancia de las mismas, el uso de inmunoglobulinas intravenosas, junto con el tratamiento con corticoesteroides e inmunosupresores, puede ser recomendable en pacientes con una recidiva de su vasculitis60.
Anticuerpo monoclonal anti-CD20 (rituximab)Los linfocitos B tienen un papel importante en la patogenia de las enfermedades autoinmunes, incluidas las vasculitis con ANCA134. Es conocido que el número de linfocitos circulantes se correlaciona con la actividad de la enfermedad.
El rituximab es un anticuerpo monoclonal contra el antígeno CD20 expresado en la superficie de los linfocitos B. Existen pocos trabajos sobre su uso en vasculitis, pero con buenos resultados135. Se ha empleado como tratamiento de inducción comparándolo con la ciclofosfamida en vasculitis ANCA positivas, y demostró no ser inferior a la ciclofosfamida incluso en pacientes con hemorragia alveolar136, y ha demostrado eficacia y pocos efectos secundarios en su uso como agente único137,138. Estos buenos datos clínicos hacen que este tratamiento represente en la actualidad una de las principales vías de estudio clínico para el tratamiento de estas enfermedades.
ConclusiónLa afectación del tracto respiratorio es frecuente en algunos tipos de vasculitis, fundamentalmente en las asociadas a ANCA. La presentación clínica, radiológica e histopatológica también es variada y condiciona la evolución. Es necesario, por tanto, una orientación clínica y diagnóstica precoz ya que, gracias a los nuevos tratamientos, y a pesar de ser enfermedades potencialmente graves, el pronóstico de estos enfermos ha mejorado de manera considerable137.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.







