


Idiopathic pleuroparenchymal fibroelastosis (IPPFE) is a very rare, recently described condition, characterised by fibrotic thickening of the pleural and subpleural parenchyma, predominantly in the upper lobes.1 Clinical manifestations and lung function tests are similar to those observed in restrictive interstitial pneumonias, and in some of the cases described, there was a history of recurrent infections, such as allergic bronchopulmonary aspergillosis or cystic fibrosis.2,3 Radiological findings include intense pleural thickening associated with signs of fibrosis, particularly in the upper lobes, with loss of volume and structural distortion, as observed in the case presented here.
This is an 82-year-old female patient with suspected usual interstitial pneumonia on CT imaging, who consulted her pulmonologist due to worsening of dyspnoea on effort and non-productive cough. The patient stated that she was a non-smoker and had not had any previous exposure to environmental allergens, radiotherapy, drugs or history of contact with asbestos. On the physical examination, the patient had a healthy appearance, with normal vital signs and 95% PaO2 (with room air). Chest examination showed reduced breath sounds and bilateral rales in the lower lobes. Heart examination showed regular frequency and rhythm, and the extremities were normal without finger clubbing. Lung function tests showed moderate restrictive ventilatory defects (FVC: 57%, FEV1: 72%). The 6-min walking test gave a distance walked of 314m (74%), no desaturation and 3–3 on the Borg scale.
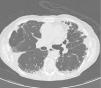
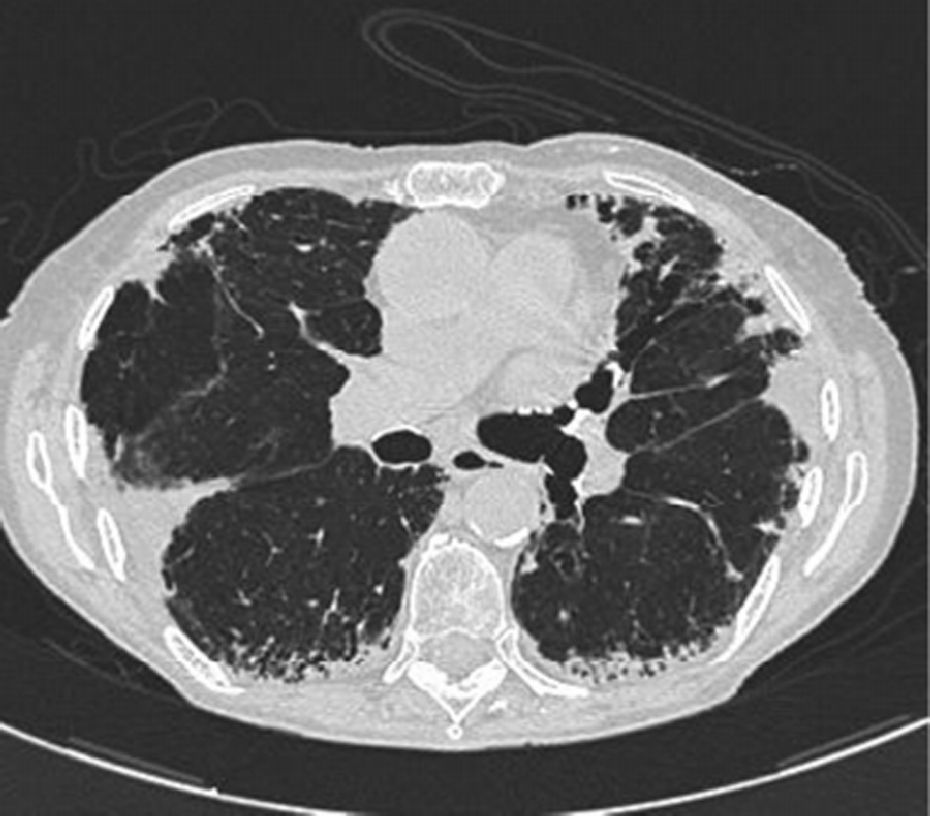
Chest X-rays showed intense apical pleural thickening and upper hilar retraction. The chest CT revealed bilateral irregular pleuroparenchymal thickening, principally in the upper and middle areas, associated with fibrotic signs (Fig. 1). Serology tests were negative for anti-scl-70, anti-Jo-1 and anti-DNA antinuclear antibodies, antineutrophil cytoplasmic antibodies, rapid plasma reagin and rheumatoid factor. A videobronchoscopy with bronchoalveolar lavage was carried out and transbronchial biopsies were obtained, giving negative cytology and microbiology results. Histopathology examination of the lung biopsy showed intraalveolar fibrosis without granulomas. The complementary imaging studies and examinations were compatible with IPPFE and the patient was initially treated with low-dose oral azathioprine and corticosteroids. After 24h, the patient showed disease progression with no response to treatment, requiring home oxygen therapy. A recent chest CT showed loss of volume and progression of pleural thickening, along with signs of fibrosis in the lower lobes. Follow-up lung function tests showed FVC 58% and FEV1 67%.
IPPFE is a very rare disease entity. Diagnosis is based on clinical, radiological and histopathological examinations. Histopathological findings include marked thickening of the visceral pleura and prominent subpleural fibrosis, with elastosis of alveolar walls. Reddy et al. describe the “definite” characteristic of IPPFE as pleural fibrosis in the upper lobes, associated with intraalveolar fibrosis accompanied by alveolar elastosis. They considered the presence of intraalveolar fibrosis as “consistent” with IPPFE but when (a) not associated with pleural fibrosis; (b) not located predominantly below the pleura; or (c) not located in the upper lobes.4,5 The treatment of IPPFE has not been determined. Kusagaya et al. described 5 cases of Japanese patients who did not receive any type of treatment with a mean follow-up of 45.2 months (7–83 months), all of who remained alive, but with clinical and functional deficit.3 In another series of 12 European patients, 9 were treated with low-dose corticosteroids, immunosuppressive drugs or N-acetylcysteine. Five (5) of these patients died within a period of 4–24 months after diagnosis.4
In conclusion, IPPFE is a very rare entity and our case is the first description of the disease in Latin America. Identification of this disorder is very important for defining its prognosis and promoting the development of alternative treatments.
Please cite this article as: Labarca G, Cabello H, Fernández Bussy S, Cabello F, Díaz JC. Un caso de neumopatía intersticial con engrosamiento pleural apical: fibroelastosis pleuroparequimatosa idiopática. Arch Bronconeumol. 2014;50:48–49.
All authors approved the final manuscript.




www.publicationethics.org.




Archivos de Bronconeumología follows the Recommendations for the Conduct, Reporting, Editing and Publication of Scholarly Work in Medical Journals