





Mycoses are serious diseases with potentially fatal outcome. The introduction of immunosuppressive treatments and life support techniques has led to a growing prevalence of different degrees of immunosuppression. Compromised immune response is the primary risk factor for the development of opportunistic mycoses. Early diagnosis and treatment are crucial for improving prognosis. However, isolation in cultures or identification using antigen detection techniques cannot distinguish between colonization and invasive infection, and the clinical status of the patient often prevents biopsy sampling. Clinicians thus find themselves in an uncertain position, requiring them to quickly recognize clinical and radiological signs and interpret microbiological results in context. The aim of this review is to provide a general overview of the profile of patients susceptible to these infections, the role of the immune system and, in more detail, the major diagnostic developments that have gained most acceptance and recognition among the scientific community.
Las micosis son enfermedades graves y potencialmente letales. Con el desarrollo de terapias inmunosupresoras y técnicas de soporte vital, la inmunosupresión en sus diferentes grados es cada vez más prevalente. El deterioro de la respuesta inmune es el factor de riesgo principal para el desarrollo de las micosis oportunistas. El diagnóstico y tratamiento precoces son factores cruciales para mejorar el pronóstico de estas enfermedades. Sin embargo, los aislamientos mediante cultivos o las técnicas de detección antigénicas no son capaces de distinguir entre colonización e infección invasiva, y las biopsias rara vez se pueden realizar por la situación clínica. Ello sitúa al médico en una situación de incertidumbre en la que debe reconocer precozmente los signos clínicos y radiológicos e interpretar los resultados microbiológicos en su contexto. El objetivo de esta revisión es aportar una visión general del perfil de paciente que sufre estas infecciones, el papel de su sistema inmune, y de forma más detallada, los principales avances diagnósticos más reconocidos y recomendados por la comunidad científica.
Fungi form a huge kingdom of eukaryotes, of which only a few are pathogenic. This review focuses on fungi which can cause respiratory disease by tissue invasion in situations of immunosuppression. Thus, diseases resulting from a disordered immune response (e.g. allergic bronchopulmonary aspergillosis) and endemic mycoses (less common and less dependent of the immune status) are not considered in this review.
Opportunistic mycoses are caused by ubiquitous fungi, including those present in commensal flora. Mycotic invasion is controlled by intact skin and mucous membranes, neutrophil activity, and cell-mediated immune response led by CD4 cells and macrophages. There is no common response to all fungi, and distinguishing clinical features have been identified: low CD4 levels are associated with pathogens such as Pneumocystis and Cryptococcus, while neutropenia can lead to Aspergillus and Mucoralean infections.
Types of immunosuppression associated with these mycoses are listed in Table 1. It is worth noting that many of the risk factors mentioned are associated with the chronic use of high-dose corticosteroids, underlining the need for careful adjustment of immunosuppression and consideration of therapeutic alternatives.
Risks Factors for Opportunistic Mycosis.
| • Disrupted natural barriers: mucositis, venous access lines, surgical wounds, intubation |
| • Prolonged neutropenia |
| • Blood cancers, whether associated or not associated with stem cell transplantation or graft-versus-host disease |
| • Disseminated malignant solid tumors |
| • Solid organ transplants |
| • HIV infection, especially with CD4 counts<200cells/mm3 |
| • Chronic lung diseases |
| • Connective tissue disease |
| • Immunosuppressive drugs, primarily corticosteroids. Other immunosuppressants associated with anti-TNFα drugs and alemtuzumab |
Clinical manifestations of pulmonary mycoses are not specific, and while it is easy to suspect mycosis in the context of known immunosuppression, the opposite approach must not be forgotten: a diffuse, slowly progressing pulmonary syndrome should alert to a possible fungal infection and lead to an investigation of any possible underlying immunosuppression. Detailed history and examination may guide diagnosis: involvement of paranasal sinuses may be suggestive of Mucor, skin lesions in disseminated disease may be a good source for biopsy, etc.
With regard to diagnosis, chest X-ray may be normal, so computed tomography is the imaging test of choice. Diffuse bilateral infiltrates with areas of ground glass and/or multimodal patterns are common, but not specific,1 and microbiological culture or the detection of fungal antigens may be positive in a colonization setting, reducing their diagnostic value for invasive disease. The yield of serological testing in immunocompromised patients is poor. Biopsy has been conventionally considered as the unequivocal test for demonstrating invasive fungal proliferation, but in many cases, the clinical status of the patient prevents sampling. Moreover, the extreme seriousness of these diseases means that appropriate empirical treatment must be started as soon as possible.
The European Organization of Research and Treatment of Cancer/Mycoses Study Group (EORTC/MSG) has defined invasive mycoses as: proven (consistent histological results or positive culture in sterile medium); probable (lower grade microbiological evidence in a patient with risk factors and consistent clinical syndrome); and possible (patients with clinical syndrome and risk factors, despite absence of microbiological confirmation).2 These guidelines reflect the effort made in unifying criteria to assist the clinician in dealing with diagnostic uncertainty.
It is important to point out that treatment does not depend exclusively on the administration of antifungals. Immunosuppression must also be addressed and reduced or reversed to achieve a level of balance that remains poorly defined. In a situation of irreversible immunosuppression, there will be little response to antifungal treatment, while recovery of the immune response may be associated with clinical worsening, due to the inflammatory response. This phenomenon is well established in HIV patients, but has also been described in non-HIV patients.3
AspergillosisAspergillosis is the primary pulmonary mycosis among critically ill patients,4 the most common species being A. fumigatus, A. flavus, A. niger and A. terreus. They are acquired by inhalation of conidia, which in an immunocompetent individual are eradicated by alveolar macrophages and neutrophils. Disease develops when this line of defense breaks down, or, more rarely, in situations of excessive inhalation of conidia, such as occurs during landslides or great catastrophes.5 Several clinical pulmonary forms have been described, the most important of which are invasive pulmonary aspergillosis (IPA), chronic aspergillosis, and aspergilloma.6 The type and severity of aspergillosis is determined by the characteristics of the patient (Fig. 1). Severe, generalized immunosuppressive states, such as prolonged neutropenia, are associated with acute invasive disease, while moderate, localized immunosuppression, such as preexisting cavities, favors the development of forms such as aspergilloma.
Invasive Pulmonary Aspergillosis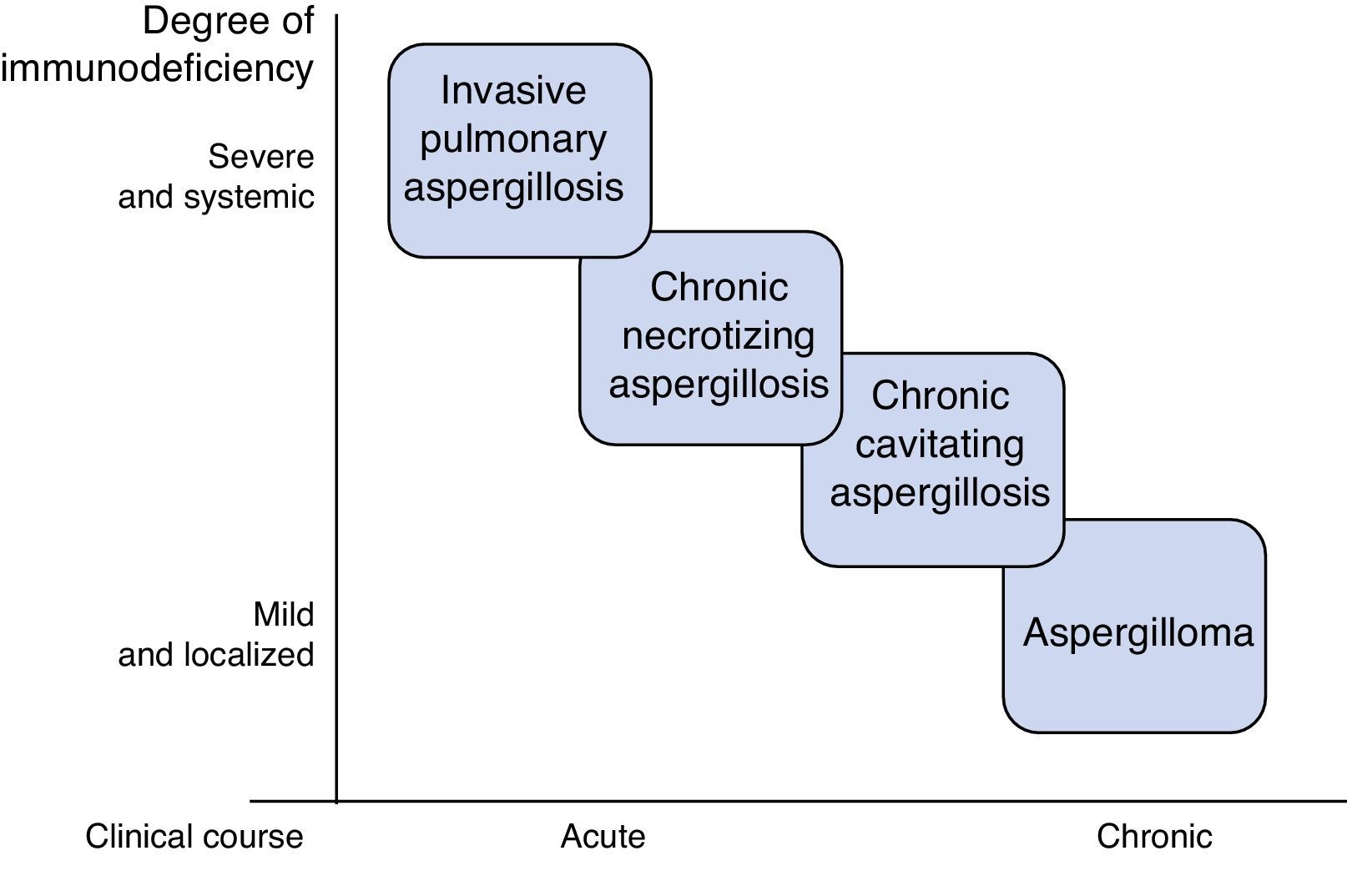
IPA is the most severe form of aspergillosis, with a mortality of around 50%. It is caused by massive proliferation of Aspergillus, with tissue invasion and high vascular tropism, promoting ischemia and dissemination.
Serious risk factors include prolonged neutropenia in patients with hematological malignancies, hematopoietic stem cell (HSC) and solid organ transplants, particularly lung and heart. Other factors thought to be intermediate include intensive care unit admission, chronic obstructive lung disease (COPD) treated with inhaled or systemic corticosteroids, chemotherapy and radiation therapy, AIDS, etc.5,7 Despite the varying importance of each risk factor, IPA in critically ill patients mostly occurs in association with COPD and prolonged corticosteroid use,4 due to their higher prevalence.
IPA progresses with fever, cough, expectoration, hemoptysis, dyspnea, and pleuritic pain. Tracheobronchial involvement may be observed, particularly in lung transplantation.8 If it disseminates, the skin, central nervous system (CNS), liver and kidneys may be affected.5
CT reveals areas of ground glass opacities with multiple nodules and cavitated lesions. Nodular lesions may be associated with perinodular hemorrhage, producing the typical “halo” sign. Subsequently, as the disease progresses, peripheral necrosis and cavitation with the air crescent sign, or necrosis and central attenuation with the reversed halo sign may develop. These radiological signs, typically related with IPA, may be imitated by other mycoses and even other non-fungal infections. Moreover, they are absent in many patients, particularly those receiving corticosteroid treatment without neutropenia.9,10
Sputum or bronchoalveolar lavage (BAL) cultures have certain limitations: sensitivity is low (cultures are negative in up to 50% of IPAs) and specificity is suboptimal, as no distinction can be made between invasive infection and possible colonization. Indeed, 30%–60% of positive cultures from lung transplant patients are thought to be due to colonization. Blood cultures are generally negative, even in disseminated forms of the disease, so yield is low.4,11
Different diagnostic techniques have been proposed to improve sensitivity and to reduce wait time for cultures. Detection of serum galactomannan, a component of the Aspergillus cell wall, in patients with hematological malignancies has a sensitivity of 71% and a specificity of 89%,12 but the yield is lower in transplant recipients and COPD patients.13,14 Galactomannan determination in BAL is more sensitive and specific than in blood in both hematological cancer patients and other groups.15 However, false positives related with betalactam antibiotics, Bifidobacterium colonization and presence of histoplasmosis, blastomycosis or penicillinosis have been reported.16,17
Another Aspergillus cell wall antigen is 1,3-β-d-glucan, and detection in serum yields similar results to galactomannan. However, it is present in the structure of most fungi, including Candida and Pneumocystis, so it is less specific and returns positive results for other invasive mycoses.17 Finally, the use of polymerase chain reaction (PCR) techniques in BAL has been proposed, resulting in sensitivity and specificity of 91% and 92%, respectively.18 The definitive test is biopsy, but in many cases sampling is not feasible, due to the clinical instability of the patient.
Delay in treatment is associated with higher mortality.19 Accordingly, if suspicion is well-grounded, treatment should be initiated. The antifungal agent of choice is voriconazole, while liposomal amphotericin B is another alternative. Second line options include posaconazole, caspofungin or micafungin. Treatment duration is at least 12 weeks, and the regimen should be tailored according to resolution and prolonged stabilization of lesions. Monitoring of galactomannan levels in blood may be useful. Surgery is limited to debridement of areas of superficial necrosis and invasion of pericardium or great vessels due to contiguity. Granulocyte stimulating factors are used in cases of neutropenia, but there is no evidence of any reduction in mortality.7,10,20,21
AspergillomaAspergilloma is a fungus ball composed of hyphae, fibrin, mucus, and cell detritus, which develops within a preexisting lung cavity caused by tuberculosis, cancer, etc. It is usually clinically asymptomatic, or presents with local symptoms limited to cough and/or hemoptysis that can be severe. It can be seen on CT, and is usually isolated and unchanging. Culture of respiratory samples and Aspergillus antibody detection (precipitins) are often positive. In the asymptomatic presentation, monitoring is recommended, while the treatment of choice for symptomatic forms is surgical resection. Oral or intralesional antifungals have not shown long-term efficacy, and their use is restricted to symptomatic cases that are not candidates for surgery.5,10,22,23
Chronic Pulmonary AspergillosisChronic pulmonary aspergillosis comprises a wide spectrum of clinical signs and symptoms due to chronic or subacute Aspergillus infection. It is caused by local fungal growth, predominantly in the lower lobes, and dissemination is exceptional. The main risk factor is chronic lung disease that generates some degree of local immunosuppression: tuberculosis, other mycobacterial infections, asthma and COPD treated with corticosteroids, lung cancer and bullae. It is associated with mannose-binding lectin polymorphisms, surfactant deficiency, and low interferon production.22,23
The most widely accepted classification subdivides chronic pulmonary aspergillosis into 3 forms: cavitary, necrotizing, and fibrosing.24 The clinical subtype is determined by the characteristics of the patient and the efficiency of their immune system.
- A.
Chronic cavitary aspergillosis: this a progressive syndrome, characterized by the development of multiple cavitated lung nodules. The clinical course is chronic, lasting months, and the most important general symptoms are weight loss, general malaise, night sweats, low-grade fever or fever. Respiratory symptoms are few and non-specific: productive chronic cough, possible hemoptysis, and dyspnea. Multinodular involvement is observed on CT, with progressive thick-walled cavities, in which aspergillomas can form.23,25
- B.
Chronic necrotizing aspergillosis: some authors consider this as a subacute form of IPA. It occurs in more highly immunosuppressed patients, and is characterized by more rapid and aggressive progression than the cavitary form, with greater invasion and tissue damage. In addition to general symptoms, fever is more frequent and respiratory symptoms more florid. CT shows patchy involvement with areas of necrosis; cavity walls are usually thinner, and the air crescent sign typical of IPA can be observed.23,25
- C.
Chronic fibrosing aspergillosis: late development of the cavitary, or more rarely, the necrotizing form. Imaging tests show progressive fibrosis, impacting on lung function tests.
Diagnostic suspicion is based on clinical and radiological findings. Laboratory reports show elevated inflammatory reactants, and precipitins are positive in most cases. Anti-Aspergillus IgE can be detected in the absence of evidence of allergic bronchopulmonary aspergillosis. Sputum culture is positive in around 40%–50% of cases, but the yield of PCR or galactomannan detection in respiratory samples is higher. Galactomannan may be elevated in blood (although not as often as in IPA), particularly in the necrotizing forms.
If clinical and radiological findings are consistent, the presence of precipitins or positive cultures is very suggestive of this clinical entity. However, some patients have negative microbiological results; moreover, intercurrent or imitative processes (mycobacterial infection, in particular) may be present, so a bronchoscopy is often essential for obtaining samples with a better yield. Definitive diagnosis is obtained from biopsy showing hyphae, local necrosis and/or granulomatous reaction. Angioinvasion is generally absent, and if it does occur, it is more often in the necrotizing form.22,23,25
Chronic forms of aspergillosis are treated with oral azoles (itraconazole or voriconazole), administered long-term or even indefinitely, due to the high risk of recurrence. Alternatives include posaconazole or amphotericin B in severe cases. Optimization of immunity is also recommended, if possible. Isolated cases of improvement with interferon have been reported in the literature.23,25 Treatment may be monitored with clinical and radiological surveillance and determination of precipitin titers.10,21 Despite treatment, mid- to long-term control of the infection is often suboptimal.
The use of primary aspergillosis prophylaxis is under debate. The main indications are listed in Table 2.26–28 It is not recommended in patients with hematological malignancies and neutropenia expected to last less than 7 days, or solid tumors receiving conventional chemotherapy. The most widely recommended drug in the different guidelines is posaconazole, as it has been most studied in clinical trials and has the advantage of being active against Mucorales fungi. Voriconazole is a reasonable alternative.
Principal Indications for Primary Aspergillosis Prophylaxis.
| Cancer patients |
| Acute myeloid leukemia or intensively-treated myelodysplastic syndrome |
| Allogenic stem cell transplantation with predicted prolonged neutropenia (more than 2 weeks) |
| Graft-versus-host disease requiring high-dose corticosteroids |
| Lung transplantation |
| Colonization before transplantation |
| Colonization within 1 year pre-transplantation |
| Presence of at least 2 of the following risk factors: |
| Induction with alemtuzumab or thymoglobulin |
| Single-lung transplantation |
| Colonization subsequent to cytomegalovirus infection |
| Rejection and increased immunosuppression |
| Acquired hypogammaglobulinemia (IgG less than 400mg/dl) |
In lung transplant recipients, evidence for the different recommendations for prophylaxis is low grade, due to the lack of appropriate studies.29,30 In standard practice, the tendency is to administer universal antifungal prophylaxis.31 Correct prophylactic dosing has not been well defined, but azoles with anti-Aspergillosis activity (itraconazole, voriconazole or posaconazole) and/or inhaled amphotericin B are generally used.
MucormycosisMucormycosis is disease caused by Mucorales fungi, the most commonly involved genera being Rhizopus, Mucor and Rhizomucor.
They are acquired by inhalation or via damaged skin and mucous membranes. They cause rapid vascular invasion, thrombosis and secondary necrosis.32 The risk factors for mucormycosis are similar to those of aspergillosis, but diabetes mellitus with poor metabolic control, iron overload, and the use of desferoxamine should also be taken into account. Controversy persists on whether voriconazole prophylaxis can increase the risk of mucormycosis32–35; the reason for this association has not been clarified, but it is posited that it may be due to Aspergillosis being displaced, allowing the positive selection of Mucorales, which are mostly resistant to voriconazole. Paradoxically, it is rare in HIV, except in cases associated with the use of intravenous drugs and corticosteroid therapy.32
The most common sites of infection are the paranasal sinuses, the lungs and the skin. Rhino-orbital mucormycosis is mostly seen in diabetics, while in hematological patients the predominant forms are pulmonary. Lung involvement behaves similarly to very severe and rapidly invasive micronodular interstitial pulmonary disease. Mortality is around 70%–75%.32,36 Clinical symptoms include dyspnea, cough, chest pain, and hemoptysis, which on occasions can be massive. It also has a high capacity for causing cavitation and progressive invasion, which may affect the pericardium, pleura, mediastinum, and chest wall. Similarly to Aspergillosis, endobronchial involvement and airway obstruction may also occur.20 Radiological findings are very varied and include infiltrates, nodules, cavities, atelectasis, pleural effusion, and mediastinal lymphadenopathies. Like Aspergillus, it produces the halo sign, and more characteristically, the reverse halo sign.32,37
The presentation, then, is similar to that of IPA, but some factors guiding the diagnosis of mucormycosis are the following: greater rhinosinusal, palatine or facial involvement, or the presence of thoracic subcutaneous tissue.38 Radiological findings of more than 10 nodules and pleural effusion are factors more often associated with mucormycosis than IPA.39 Moreover, galactomannan and 1,3-β-d-glucan are negative in this entity. A history of voriconazole use and lack of response to this drug support the suspicion. None of these findings is specific, but are useful as guidance when waiting for microbiological results.
Diagnosis is made from respiratory cultures or biopsies. Since its presence in respiratory samples is rare, isolation in a consistent clinical context is considered diagnostic. Blood cultures are negative, even in invasive forms.37,38
Treatment must be initiated promptly,40,41 with resolution of risk factors, surgical debridement of accessible areas and administration of antifungal agents.32 Only amphotericin B and posaconazole are known to be effective. Amphotericin B is the first line treatment, while posaconazole is used in cases of renal dysfunction and in sequential oral treatment in the maintenance phase. Duration of treatment has not been established and should be individualized according to clinical response and resolution of lesions.20,21,38,42,43 Surgery plays a fundamental role in the resolution of local forms. The effectiveness of granulocyte stimulating factors in neutropenic patients has been defined.42,44
CryptococcosisThe most important pathogens of the Cryptococcus genus are Cryptococcus neoformans (C. neoformans) and the emerging Cryptococcus gattii (C. gattii). They are ubiquitous and are isolated from soils, particularly in areas frequented by birds and bats.45 While the distribution of C. neoformans is universal, C. gattii is more common in subtropical regions, and its presence has increased in recent years in some areas of the North Pacific.46
The invasive capacity of Cryptococcus is due to its expression of a polysaccharide capsule with antiphagocytic properties. It enters by the airways and causes 2 distinct clinical syndromes: meningitis and meningoencephalitis. Its pulmonary transit, however, can be blocked by a cell-mediated immune reaction and the development of granulomas; in an immunocompromised host, invasion can occur.47–49
The major risk factor is HIV infection, particularly when CD4 counts are below 100cells/mm3, CNS involvement being the predominant clinical manifestation.50 In other immunosuppressive states, such as chronic lung diseases, patients on long-term corticosteroids, or solid organ transplant recipients, the resulting syndrome is predominantly respiratory.21,51C. gattii affects the lungs, even in immunocompetent patients.46
Pulmonary cryptococcosis is characterized by fever, with dyspnea, dry cough, night sweats, and progressive, occasionally fulminant, respiratory failure.47,52,53 Severe pulmonary presentations involve a risk of dissemination to the CNS and to other regions, particularly the skin.54
The most common radiological pattern is multiple nodular involvement. Reticulonodular forms are more typical of invasive syndromes. Involvement in the form of lobar or segmentary consolidation is also possible. Thoracic lymphadenopathies may also occur. Pleural effusion and cavitation are exceptional.55
Cryptococcus in colonizations is unusual; therefore, isolation in a sputum or BAL culture consistent with clinical signs is sufficient for diagnosis. Cryptococcus antigen determination in plasma is very useful, but it can be negative in some limited pulmonary forms.53,56 Antigen detection in sputum or BAL is unreliable.47 Cerebrospinal fluid (CSF) can be studied by India ink staining, antigen detection and/or culture. Routine lumbar puncture is not recommended for all pulmonary forms, but rather in the presence of neurological symptoms, severe immunosuppression, signs of dissemination, or high serum antigen levels.57
The treatment of pulmonary forms has not been widely studied, and the principal recommendations are based on the experience of treatment of HIV patients with CNS involvement. For mild or moderate respiratory syndromes, fluconazole for 6–12 months is recommended.20,21,47,58 In severe manifestations or CNS involvement, at least 2 weeks combined treatment with amphotericin B and flucytosine is recommended.21,58,59 This treatment should be followed sequentially by a 6–12 month course of fluconazole. Treatment duration must be individualized on the basis of risk factors and recovery of immunosuppression status. For example, in HIV subjects, maintenance treatment should continue until viral replication is controlled and a stable CD4 count>100cells/mm3 has been achieved. Moreover, initiation of antiretroviral treatment should be delayed for at least 4–8 weeks to avoid immune reconstitution inflammatory syndrome, underlining once more the difficulties in achieving the immunological balance required in the management of opportunistic mycosis.56,60
PneumocystosisPneumocystis jirovecii is an atypical fungus, acquired by the airborne route, even in childhood.61 The main risk factor for developing this mycosis is HIV infection, particularly when CD4 levels are below 200cells/mm3. It can, however, also occur in other cell-mediated immunosuppressive states common to other mycoses.62,63 It is important to note that the immunosuppressive agents most frequently cited are corticosteroids, particularly in association with dose increases or reductions from previously high doses.64,65
The clinical syndrome in patients with HIV infection is subacute, with fever, dry cough, and progressive dyspnea, and generalized asthenia and weight loss. However, in immunosuppressed patients without HIV infection, the clinical picture is more acute, prognosis is poorer and the rates of acute respiratory failure, need for ventilatory support and mortality are higher.64,66–69 Some authors have associated this increased mortality in non-HIV patients with more severe immunosuppression status, and with delay in diagnosis and treatment, since the suspicion of pneumocystosis is not so strong.67
Diffuse bilateral interstitial infiltrates may be seen on X-ray. CT provides a more defined image of bilateral ground glass opacities, possible pneumatoceles and pneumothorax due to local parenchymal damage. It is rare to see lobar infiltrates, nodules, cavities, or pleural effusion. If the CT is normal, the diagnosis should be questioned.70
During pneumocystosis, 1,3-β-d-glucan levels are raised, leading many authors to suggest that a positive finding in a high risk patient with consistent signs and symptoms may be sufficient to reach a diagnosis. However, in view of the association with pneumocystosis with other intercurrent processes, the recommendation is to obtain a more specific diagnosis.71,72 Since it cannot be cultured, visualization using immunofluorescence techniques is needed, and yield is high. Immunofluorescence can be used in induced sputum, with sensitivity ranging between 55% and 90%, or BAL with a sensitivity of around 90%–100%. Biopsy is rarely necessary. PCR values have yet to be defined.73
If there is a clinical suspicion of pneumocystosis, treatment must be initiated without waiting for sampling of respiratory specimens, as eradication of pathogens from the respiratory secretions will take several weeks.74,75
In HIV patients, the treatment of choice is cotrimoxazole for 21 days. Alternatives include pentamidine, dapsone, atovaquone, or combined clindamycin and primaquine, depending on severity. Co-administration of corticosteroids is recommended in severe cases.76 Antiretroviral treatment must also be delayed for at least 2 weeks.68,76,77
Treatment of non-HIV patients is less widely studied, and most of the recommendations for antimicrobials apply in these cases. Corticosteroid treatment is more controversial in these patients. Some small studies were not able to show clear efficacy in terms of mortality, so corticosteroids should be continued in patients previously receiving this treatment, and initiated in cases of severe lung inflammation.21,78,79 Cotrimoxazole is the agent of choice for secondary prophylaxis and should be administered until immunological recovery. Cotrimoxazole is also given for primary prophylaxis. Alternatives include atovaquone, dapsone, or aerosolized pentamidine. The main indications are listed in Table 3.1,27,80,81
Principal Indications for Primary Anti-Pneumocystis jirovecii Prophylaxis.
| • HIV with CD4 count<200cells/mm3 |
| • Prednisone treatment at doses>20mg/day for more than 1 month and presence of another cause of immunosuppression |
| • Alemtuzumab treatment |
| • Acute lymphoid leukemia |
| • Blood cancer patient on intensive regimen containing purine analogs |
| • Stem cell transplant recipient |
| • Solid organ transplant recipient |
| • Rheumatic disease requiring induction immunosuppressive treatment (e.g., corticosteroids and cyclophosphamide) |
The authors state that they have no conflict of interests.
Please cite this article as: Curbelo J, Galván JM, Aspa J. Actualización sobre Aspergillus, Pneumocystis y otras micosis pulmonares oportunistas. Arch Bronconeumol. 2015;51:647–653.







