


Dyskeratosis congenita or Zinsser-Cole-Engman syndrome is a rare hereditary disease with multisystemic involvement. At least 12 genes related to telomere maintenance have been implicated in the pathogenesis of the disease.
In terms of clinical symptoms, nail dystrophy, reticular pigmentation, and oral leukoplakia are the most common manifestations in this disease. Pulmonary fibrosis, although it affects only 20% of patients, causes the greatest morbidity and mortality.1
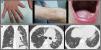
We report the case of a 49-year-old man, referred to a pulmonology outpatient clinic due to a 6-month history of dry cough. He had seasonal rhinoconjunctivitis and dust allergy and worked in a transport company in frequent contact with truck exhaust fumes. In 2000, he was diagnosed with X-linked congenital dyskeratosis by the dermatology department, with symptoms of nail dystrophy, reticular pigmentation, and oral leukoplakia (Fig. 1A). He was also being monitored by the hematology department for aplastic anemia. Karyotype analysis in peripheral blood and bone marrow was normal. Regarding family history, only one cousin on his mother's side had been diagnosed with congenital dyskeratosis. His parents were dead and his brothers had also died when they were young, cause unknown.
Lung auscultation revealed bibasal velcro crackles and resting oxygen saturation by pulse oximetry was 98%. Chest X-ray showed predominantly reticular involvement in both upper lobes and blood test results, including the autoimmunity study, were normal.
The patient showed evidence of moderate restriction and decreased carbon monoxide diffusion capacity (DLCO) in respiratory function tests (FEV1 71%; FVC 71%; FEV1/FVC 60%; TLC 66%; DLCO 62%). He also had significant desaturation during the 6-minute walk test, and covered less distance than predicted, showing moderate dyspnea at the end of the test.
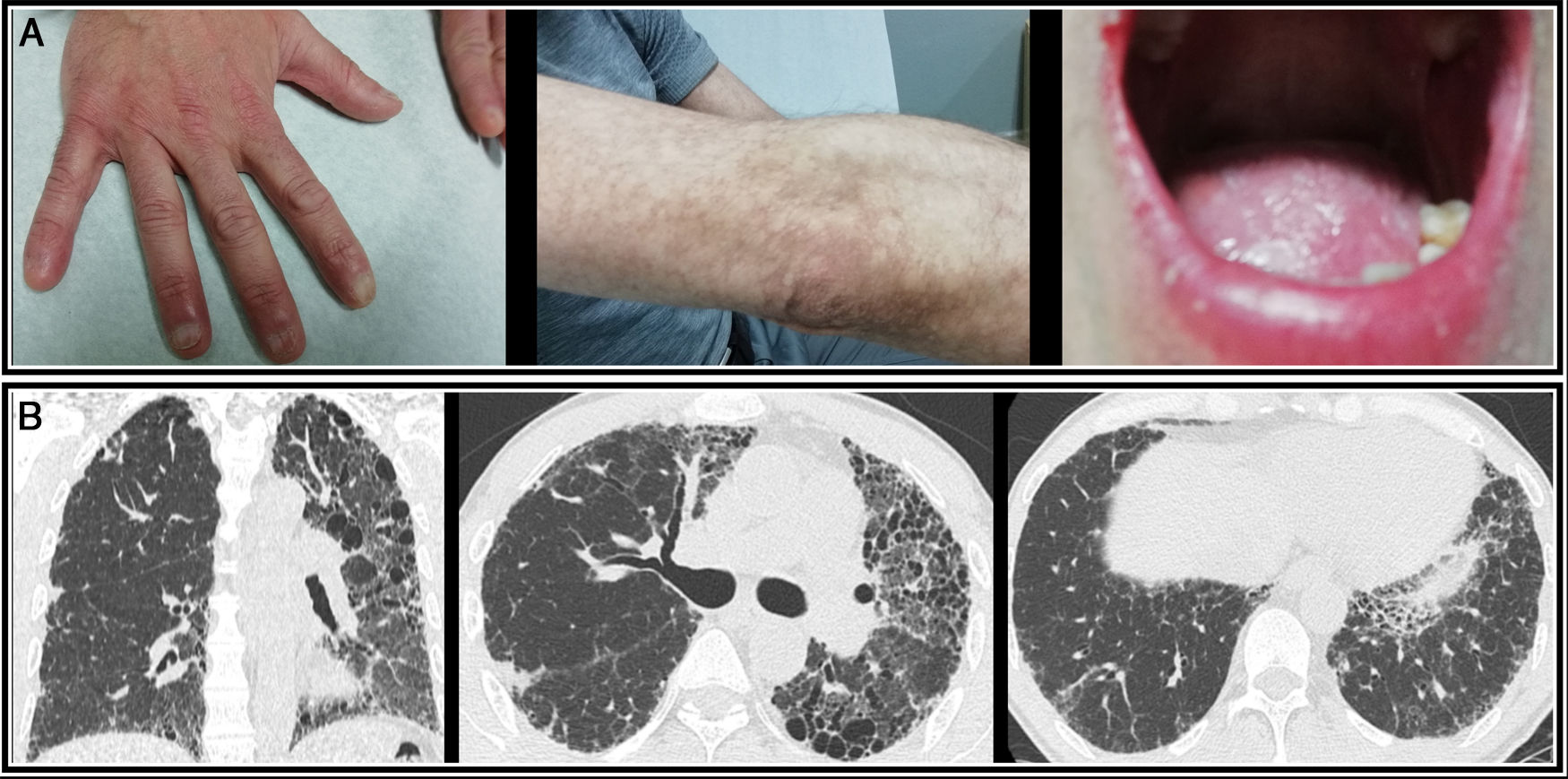
The study was extended with high-resolution computed tomography (HRCT) which showed diffuse parenchymal involvement, with increased volume reduction in the left hemitorax and reticular opacities with bronchiectasis and traction bronchiolectasis in both upper lobes and the lingula, consistent with fibrosing disease Fig. 1).
Bronchoscopy was also performed with aspiration and bronchoalveolar lavage: cytology and microbiological results were negative. Given these findings, antifibrotic treatment was started with nintedanib, which was well tolerated.
Telomeres were analyzed in the Telomeropathy Detection Department of the CSIC Institute of Biomedical Research. The study showed that the patient's telomere length was below the 10th percentile compared to the healthy population of the same age. Exon sequencing of the DKC1 gene, associated with X-DC, showed the pathogenic variant (NM_001363.4) c.203rd>G; p.H68R in exon 4 of the DKC1 gene in homocygosis. A family genetic study was not possible, as the patient's brothers and parents had died.
A diagnosis of mild-moderate fibrosing disease (GAP 3, stage I), associated with congenital dyskeratosis with hematological and cutaneous involvement and telomere shortening, was given.
In the last 3 months, the patient presented functional progression (57% FVC and 26% DLCO) and radiological progression on HRCT, with signs of honeycombing, with multiple cysts predominantly in the upper lobes and in the left lung, and an increased pulmonary artery caliber indicating pulmonary hypertension.
Given this rapid decline, with poor response to treatment, he was referred to 3 reference centers to be evaluated for lung transplantation. All centers rejected the procedure due to the high risk, given their limited experience in this type of patients, and the poor prognosis and likelihood of post-transplantation morbidity. Palliative care was intensified, particularly for the patient's disabling dyspnea. In June 2019, he died of respiratory failure.
The true prevalence of dyskeratosis congenita is unknown. It has been estimated to affect approximately 1 in 1 million of the population. Genetic variants with different degrees of penetrance and severity and 3 types of genetic inheritance have been identified: autosomal recessive, X-linked, and autosomal dominant.2
Genes associated with congenital dyskeratosis and telomere shortening to date include CTC1, ACD, NHP2, DKC1, PARN, NOP10, TERC, RTEL1, TINF2, TERT, and WRAP53. The dyskerin pseudouridine synthetase 1 (DKC1) gene is the most common (30%) and inheritance is X-linked: about 40 pathogenic variants of this gene have been described. However, the causative gene is not identified in 20%–30% of cases.3
As observed in the clinical case presented, although mucocutaneous manifestations are the most frequent, pulmonary fibrosis constitutes one of the most serious manifestations.1 It affects about 1 in 5 individuals with dyskeratosis congenita.4 It is usually diagnosed in individuals aged 20 to 40 years as a result of the study of respiratory symptoms or respiratory infection.5 In some cases, it develops after bone marrow transplantation. In others the cause is unknown.
The combination of pulmonary fibrosis and bone marrow failure is a powerful predictor of telomere dysfunction.6 Telomere shortening is described in up to 25% of cases of idiopathic pulmonary fibrosis and in more than 50% of family forms and contributes to increased epithelial apoptosis.
The mean survival of patients with associated idiopathic pulmonary fibrosis is less than 3 years from diagnosis.7 However, these patients mainly die due to complications from bone marrow failure (60%–70%).
The only treatment that has been shown to prolong survival in these patients is lung transplantation.8 However, when telomere shortening is an associated factor, prognosis is worse and post-transplant morbidity is more severe than in cases of non-familial idiopathic pulmonary fibrosis.9–11 Experience is scant, but cases with good outcomes have been reported in the literature and some authors believe that transplantation is a feasible option in these patients.2,12
Given the rapid progress of this disease, screening for telomere shortening is a very important tool for selecting patients for referral for expeditious lung transplantation.13
Please cite this article as: Guevara Velázquez V, González JM, García Arias-Salgado E, Cordovilla Pérez R, Iglesias Heras M, Hernández Mezquita MÁ, et al. Manifestación pulmonar de una enfermedad hereditaria de expresión fundamentalmente mucocutánea. Arch Bronconeumol. 2020;56:468–469.







