



Chronic obstructive pulmonary disease (COPD) and lung cancer (LC) are prevalent diseases and a leading cause of morbidity and mortality worldwide. There is strong evidence to show that COPD is an independent risk factor for LC. Chronic inflammation plays a significant pathogenic role in COPD comorbidities, particularly in LC. On the one hand, cellular and molecular inflammatory mediators promote carcinogenesis and, on the other, chronic inflammation impairs the capacity of the immune system to identify and destroy pre-malignant and malignant cells, a process known as tumor immune surveillance. This altered antitumor immunity is due in part to the expansion of myeloid-derived suppressor cells (MDSC), which are characterized by an ability to suppress the antitumor activity of T-cells by down-regulation of the T-cell receptor ζ chain (TCR ζ) through the catabolism of l-arginine. COPD and LC patients share a common pattern of expansion and activation of circulating MDSC associated with TCR ζ downregulation and impaired peripheral T-cell function. The objectives of this study were to review the evidence on the association between COPD and LC and to analyze how MDSC accumulation may alter tumor immune surveillance in COPD, and therefore, promote LC development.
La enfermedad pulmonar obstructiva crónica (EPOC) y el cáncer de pulmón (CP) son enfermedades prevalentes y representan causas principales de morbimortalidad a nivel global. Existe firme evidencia que demuestra que la EPOC es un factor de riesgo independiente de CP. La inflamación crónica juega un rol patogénico significativo en el desarrollo de las comorbilidades en la EPOC, y en particular en el CP. Diferentes mediadores inflamatorios celulares y moleculares promueven la tumorigénesis e inhiben la capacidad del sistema inmunitario de reconocer y eliminar células premalignas y malignas, proceso conocido como inmunovigilancia tumoral. Esta alteración de la inmunidad antitumoral se debe en parte a la expansión de las células mieloides supresoras (myeloid derived suppressor cells [MDSC]), que se caracterizan por suprimir la función efectora antitumoral de linfocitosT mediante la reducción de la expresión del T-cell receptor ζ (TCRζ) a través del catabolismo de la L-arginina. Los pacientes con EPOC y CP comparten un patrón similar de aumento y activación de las MDSC circulantes asociado a la reducción de la expresión del TCRζ y a la alteración de la función de los linfocitosT periféricos. Los objetivos de este artículo son revisar la evidencia sobre la asociación entre EPOC y CP, y analizar cómo la acumulación de MDSC podría alterar la inmunovigilancia tumoral en la EPOC y favorecer el desarrollo de CP.
Chronic obstructive pulmonary disease (COPD) and lung cancer (LC) are prevalent diseases and a leading cause of morbidity and mortality worldwide.1 COPD affects approximately 10% of adults worldwide,2 with a similar prevalence in Spain.3 It is estimated that by 2020, COPD will become the third leading cause of death and disability.4 LC is the major cause of cancer death in women and in men.5,6 Five-year survival is poor, ranging from 6% to 14% in men and 7% to 18% in women.7
COPD is characterized by airflow obstruction associated with chronic inflammation of the airways and the lung parenchyma caused by the inhalation of gases and noxious particles, primarily tobacco smoke. Comorbidities and exacerbations add to the severity of the disease.8,9 Clinical symptoms, progress and prognosis are crucially influenced by systemic manifestations and comorbidities, irrespective of the grade of airflow obstruction.10,11
In recent years, evidence has emerged of the pathogenic role of chronic inflammation in the development of comorbidities associated with COPD. One of the most significant, and a common cause of death in COPD patients, is LC.12–14 Chronic inflammation plays an important role in tumorigenesis: mechanisms are many and varied. It enhances tumor formation, and also disarms the tumor immune surveillance capacity of the immune system, which would normally recognize and eliminate pre-malignant and malignant cells.15–17 Phenomena associated with the mechanisms of cancer for evading antitumor immunity include the profound alteration of myelopoiesis and expansion of myeloid-derived suppressor cells (MDSC), which inhibit the main anti-tumor effectors, the T cells and natural killer (NK) cells. These phenomena are also associated with chronic non-malignant inflammatory processes, such as infectious diseases, inflammation and diseases caused by a dysregulated immune system.16,18,19 Our group and others have observed expansion and activation of circulating MDSCs associated with T cell dysfunction in COPD patients.20,21 The questions arising from these observations are the following: do COPD patients have altered tumor immune surveillance as a consequence of MDSC expansion? Would this explain, in part, the high incidence of LC in these patients? In this article, we aimed to review the studies that confirm the association between COPD and LC and to analyze how increased MDSC levels might alter tumor immune surveillance in COPD.
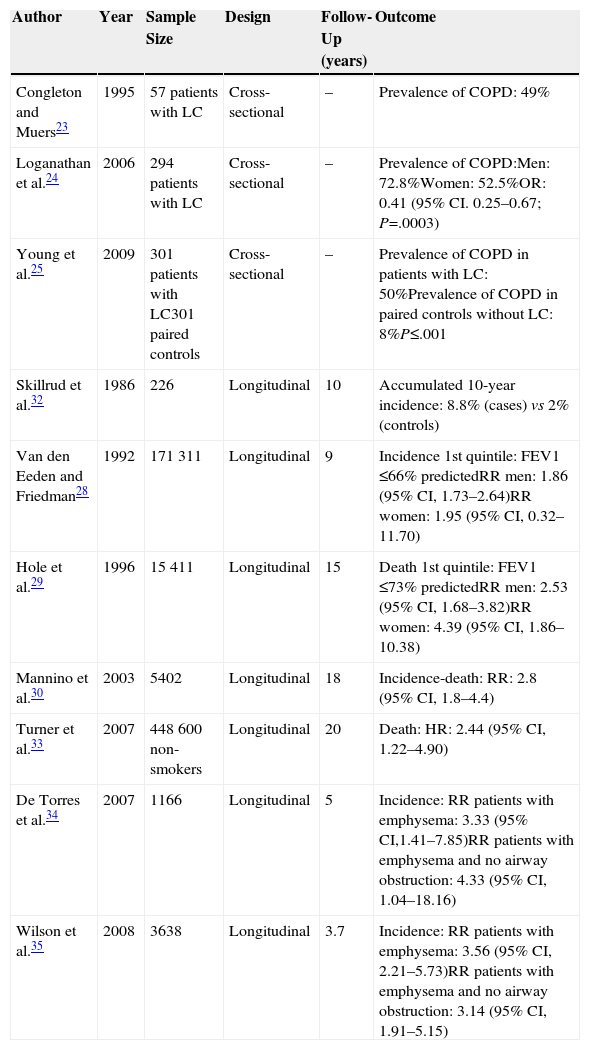
Chronic Obstructive Pulmonary Disease as a Risk Factor for Lung CancerTobacco smoke is the main risk factor for LC and COPD, but not all smokers develop these diseases. However, patients who do develop COPD have a greater risk of developing LC, irrespective of the intensity of their exposure to tobacco smoke, their age or their sex.22 Researchers have used different methodologies to confirm the association between COPD and LC (Table 1). Cross-sectional studies have reported that between 40% and 70% of patients with a diagnosis of lung cancer have COPD.23–25 According to Young et al.,25 about 50% of LC patients had concomitant COPD compared with only 8% of the non-LC control group of smokers. This points to a disproportionately large number of cases of LC in smokers with pre-existing COPD. Longitudinal prospective studies also confirmed the high incidence of LC in patients with COPD. Wasswa-Kintu et al.26 conducted a meta-analysis that included large (>5000 participants), prospective, population-based studies, adjusted for confounding factors (age, race, intensity of exposure to cigarette smoke).27–30 They analyzed a total sample of 204990 participants, of which 6185 developed or died from LC in a follow-up period of 9–18 years. Participants with poorer lung function had a greater risk of developing LC, but patients with less compromised lung function (FEV1 80%–100% predicted) also had a high risk of developing LC. This relationship was even more marked in women than in men.26 De Torres et al.31 published a smaller, non-population based study, which found that the incidence of LC was greater in patients with mild-to-moderate obstruction in baseline lung function tests. Skillrud et al.32 reported an accumulated 10-year rate of LC of 8% in subjects with FEV1 ≤70%, compared to 2% in controls. The association between chronic bronchitis and emphysema and LC mortality was also confirmed in a large population cohort of non-smokers.33
Characteristics of the Principal Studies of the Association Between Chronic Obstructive Pulmonary Disease (COPD) and Lung Cancer (LC).
| Author | Year | Sample Size | Design | Follow-Up (years) | Outcome |
|---|---|---|---|---|---|
| Congleton and Muers23 | 1995 | 57 patients with LC | Cross-sectional | – | Prevalence of COPD: 49% |
| Loganathan et al.24 | 2006 | 294 patients with LC | Cross-sectional | – | Prevalence of COPD:Men: 72.8%Women: 52.5%OR: 0.41 (95% CI. 0.25–0.67; P=.0003) |
| Young et al.25 | 2009 | 301 patients with LC301 paired controls | Cross-sectional | – | Prevalence of COPD in patients with LC: 50%Prevalence of COPD in paired controls without LC: 8%P≤.001 |
| Skillrud et al.32 | 1986 | 226 | Longitudinal | 10 | Accumulated 10-year incidence: 8.8% (cases) vs 2% (controls) |
| Van den Eeden and Friedman28 | 1992 | 171311 | Longitudinal | 9 | Incidence 1st quintile: FEV1 ≤66% predictedRR men: 1.86 (95% CI, 1.73–2.64)RR women: 1.95 (95% CI, 0.32–11.70) |
| Hole et al.29 | 1996 | 15411 | Longitudinal | 15 | Death 1st quintile: FEV1 ≤73% predictedRR men: 2.53 (95% CI, 1.68–3.82)RR women: 4.39 (95% CI, 1.86–10.38) |
| Mannino et al.30 | 2003 | 5402 | Longitudinal | 18 | Incidence-death: RR: 2.8 (95% CI, 1.8–4.4) |
| Turner et al.33 | 2007 | 448600 non-smokers | Longitudinal | 20 | Death: HR: 2.44 (95% CI, 1.22–4.90) |
| De Torres et al.34 | 2007 | 1166 | Longitudinal | 5 | Incidence: RR patients with emphysema: 3.33 (95% CI,1.41–7.85)RR patients with emphysema and no airway obstruction: 4.33 (95% CI, 1.04–18.16) |
| Wilson et al.35 | 2008 | 3638 | Longitudinal | 3.7 | Incidence: RR patients with emphysema: 3.56 (95% CI, 2.21–5.73)RR patients with emphysema and no airway obstruction: 3.14 (95% CI, 1.91–5.15) |
Other researchers used 2 longitudinal studies to explore the independent relationship between the two COPD components – airway obstruction and pulmonary emphysema – with the risk of LC. Results showed that even in the absence of airway obstruction, patients with emphysema had a substantially higher risk of LC, confirming an independent relationship between both components.34,35 Zurawska et al.36 reviewed these 2 studies and another 4 with a different design (case control studies). The relative risk (RR) for LC in the presence of “any grade of emphysema” reported by radiologists (semiquantitative analysis) was 1.9–4.9, with a total RR of 2.34 (95% CI, 1.46–3.76). A population-based prospective cohort study designed to examine the relationship between systemic inflammation markers and COPD comorbidities found that simultaneous elevations of C-reactive protein (CRP), fibrinogen, and leukocytes were associated with a 2–4 fold risk of comorbidities, including LC.37 All this evidence suggests that chronic inflammation plays a significant role as a pathogenic link between COPD and LC.12,13
Immune Editing and Tumor Immune SurveillanceOver a century ago, Paul Erlich38 proposed the idea that the immune system is capable of eliminating tumor cells. The concept that the immune system controls cancer did not emerge again until later, when it was better understood and the existence of tumor antigens had been demonstrated.39 Burnet and Thomas introduced the hypothesis of tumor immune surveillance, by which the immune system of an immunocompetent host inhibits the development of cancer by acting as an extrinsic tumor suppressant.15,17,40–43 Pre-malignant and malignant cells express specific antigens (tumor-specific antigenic molecules) or tumor-associated antigens (molecules expressed differently by tumor cells and normal cells). Antigen-presenting cells (APC) can recognize and process these tumor antigens on the malignant cells and present them to T cells for elimination.44,45
However, tumor immune surveillance is not always enough to prevent some malignant cells from escaping the immune system and developing tumors which coexist with an active immune system that can even, paradoxically, enhance their growth and modulate their immunogenicity (tumor editing).43,46,47 The notion that the immune system can on the one hand protect the host from developing cancer, but on the other, mold the tumor's immunogenicity and even promote its growth forms the basis of the most complex and comprehensive hypothesis of the intricate relationship between cancer and immunity: cancer immunoediting. This new concept comprises 3 sequential phases: elimination, equilibrium, and escape43,47 (Fig. 1).

The concept of cancer immunoediting describes the relationship between the immune system and transformed, malignant cells. It consists of 3 phases: tumor elimination or immune surveillance, equilibrium and escape. For a more detailed explanation, see the text.
In the elimination phase, previously called tumor immune surveillance, the innate and the adaptive immune systems detect and destroy transformed cells long before the tumor is clinically detectable. Tumor cells induce early mediators that act as “danger signals”, such as type I interferons. These cytokines activate the APCs and promote CD4+ helper and CD8+ cytotoxic T cells, the main effectors of adaptive antitumor immunity.43 Cell stress molecules expressed on the surface of the tumor cells bind to cell receptors which activate innate immunity, such as macrophages and NK cells, releasing cytolytic molecules.43
If the transformed cells are fully eliminated, the tumor does not develop in the host, but if elimination is incomplete, the intact tumor cells begin an equilibrium phase, during which the adaptive immune system prevents tumor growth by maintaining the residual tumor cells in a state of dormancy. Cells can remain in this state for decades without causing disease in the host. During this phase, the constant selective pressure of adaptive immunity on genetically unstable cells causes “edited” cell variants to emerge. These variants are less immunogenic, and thus are less sensitive to immune detectors and effectors, and use a wide variety of mechanisms associated with the chronic inflammatory process caused by the cancer itself, known as tumor escape mechanisms, to induce a local and systemic state of antitumor immunosuppression. In the escape phase, the immune system cannot block tumor proliferation, progression, and dissemination, so the cancer transforms into clinically detectable disease.15,43,47 Tumors use a wide range of complex strategies to establish a local and systemic environment of immune privilege. These include alteration of the mechanisms of presentation of tumor antigens, production of immunosuppressive factors, such as transforming growth factor β (TGF-β) and soluble FAS ligand, regulatory T cell (Treg) expansion, proximal signaling defects in the T cell receptor (TCR) due to down-regulation of zeta subunit (TCR ζ) expression and profound alteration of myelopoiesis, causing APC defects (particularly of the dendritic cells), and MDSC accumulation.18,48,49
Chronic Inflammation and Immunosuppression: The Role of Myeloid-Derived Suppressor CellsSeveral non-malignant etiological processes, such as infections, inflammatory and autoimmune diseases, like cancer, show a pattern of chronic inflammation associated with immunosuppression. This phenomenon is mediated in part by the MDSCs.18,50 The role of MDSCs in respiratory diseases, including asthma, pneumonia, and cystic fibrosis, has recently come under scrutiny.51–54 MDSCs comprise a morphologically heterogeneous population of cells produced in the bone marrow. They occur in various states of differentiation and maturation and are characterized by their ability to inhibit the function of different components of the innate and adaptive immune system, such as NK cells, CD4+ and CD8+ T cells, dendritic cells and macrophages.18 They were first described, 40 years ago, as “natural suppressors” in animal tumor models, in which they were associated with the dysfunction of cytotoxic lymphocytes.55 In the inflammatory and malignant disease states described above, cytokines and soluble factors, such as GM-CSF, G-CSF, M-CSF, SCF, INF-γ, IL-1β, VEGF and IL-3, are released. This phenomenon affects myelopoiesis by increasing the proliferation of myeloid precursors and partially blocking their differentiation. MDSCs accumulate and subsequently migrate to secondary lymphoid organs and tissues, where they act as immune suppressors. MDSCs use a variety of mechanisms to modulate immune response, including depletion of l-arginine, an amino acid nutrient critical to T cell functioning. To achieve this, MDSCs release arginase I (ARG I), an l-arginine-degrading enzyme, into the tissue microenvironment and the bloodstream. l-Arginine deprivation reduces the expression of TCR ζ, preventing the T-cell proliferation and production of effector cytokines56–58 (Fig. 2).

MDSCs use a variety of mechanisms to suppress the function of innate and adaptive immunity function of cells, including the release of ARG I and the reduction of TCR ζ in CD4, CD8 and NK T cells. For a more detailed explanation, see the text.
TCRs are complex molecules on the T cell membrane that play a key role in the process of antigen recognition, intracellular signal transduction, transcription, and expression of effector molecules in the context of antigen presentation by APCs.59 Reduced TCR ζ expression affects the normal functioning of the T cell by blocking signal transduction between the TCR and transcription and gene expression factors, manifesting in lower lymphocyte proliferation and reduced production of effector molecules. Various chronic inflammatory conditions, including cancer, chronic infections (e.g., HIV, active tuberculosis, leprosy), and autoimmune diseases (e.g., systemic lupus erythematosus and rheumatoid arthritis), are characterized by a reduction in specific TCR ζ expression, associated with defective T cell function.50 The same phenomenon has recently been reported in pulmonary CD8+ T cells in COPD patients.60 Studies conducted both in vitro and in vivo in chronic inflammation animal models have shown that MDSCs modulate the function of T cells by reducing TCR ζ expression, causing immunosuppression.60–64 MDSC expansion and activation and reduced TCR ζ expression, are typical features of non-malignant diseases which share the common denominator of a chronically stimulated immune system. This observation has led to the interpretation that both are protection mechanisms deployed by the host for lessening potential tissue damage from a persistently stimulated immune system.18,50 This host protection mechanism is manipulated and used by tumor cells in order to escape and evade immunity.47,48,50,55
Myeloid-Derived Suppressor Cells Expansion in Chronic Obstructive Lung Disease: Potential Tumor Immune Surveillance ChangesSystemic inflammation associated with COPD is defined as high levels of various biomarkers, including leukocytes, CRP, fibrinogen, interleukin-6 (IL-6), interleukin-8 (IL-8) and tumor necrosis factor-alpha (TNFα).8,65
MDSCs occur as a subtype of regulatory cells in a wide variety of non-malignant chronic inflammatory diseases, and our group and others have shown that they participate in COPD pathogenesis. Our study revealed an increase in circulating MDSCs in COPD patients associated with increased serum levels of ARG I enzyme and reduced TCR ζ expression (Fig. 3).20 Kalathil et al.21 reported peripheral MDSC accumulation, increased levels of functional suppressor Tregs, and T cell dysfunction in COPD patients. These observations show an MDSC expansion pattern similar to that described in other chronic inflammatory diseases of various etiologies, supporting the notion that MSDCs naturally regulate a persistently activated immune system.18,50 These results suggest that the adaptive immunity of COPD patients may be chronically suppressed, and as a consequence, unable to eliminate pre-malignant and malignant cells in the early stages of tumorigenesis, as would normally occur in the elimination or tumor immune surveillance phase.16,43,47
 Box plot of MDSC (expressed as % of mononuclear cells). (B) Mean (±MSD) of TCR ζ in non-smokers, smokers with normal lung function, and smokers with COPD.")
(A) Box plot of MDSC (expressed as % of mononuclear cells). (B) Mean (±MSD) of TCR ζ in non-smokers, smokers with normal lung function, and smokers with COPD.
These observations led us to hypothesize in a second study that this MSDC expansion pattern associated with reduced TCR ζ might contribute to the increased incidence of LC in COPD patients. We compared these same variables in COPD patients (with and without LC), smokers with normal lung function (with and without LC), and in non-smokers without LC. This study confirmed that the percentages of circulating MDSCs and ARG I serum levels are similarly elevated in patients with LC, COPD, and both LC and COPD and TCR ζ expression is similarly reduced. These findings support our working hypothesis and suggest that the mechanisms orchestrated by the cancer for evading the immune system (tumor escape phase) are already present in COPD patients, and may alter tumor immune surveillance, thus contributing to the increase in the incidence of LC in these patients.66
ConclusionsMDSC expansion, associated with chronic inflammation in COPD, may play a pathogenic role in tumor formation, by causing T cell dysfunction and by inhibiting tumor immune surveillance, thus contributing to the increased risk of LC in COPD patients.
FundingFunding was received from the General Department of Research and Technological Development and Innovation of the Ministry of Innovation, Interior and Justice of the Autonomous Community of the Balearic Islands, and FEDER Funds. Competitive Groups (PRE-R-22528-2011).
Conflict of InterestsThe authors state that they have no conflict of interests.
Please cite this article as: Scrimini S, Pons J, Sauleda J. Células mieloides supresoras: potencial vínculo entre la enfermedad pulmonar obstructiva crónica y el cáncer de pulmón. Arch Bronconeumol. 2016;52:29–35.







