


La discinesia ciliar primaria (DCP) es una enfermedad caracterizada por una alteración en la estructura ciliar que impide el correcto aclaramiento de las secreciones respiratorias. Su diagnóstico es complejo y se basa en una combinación de técnicas. El objetivo de este estudio fue diseñar un panel de genes incluyendo todos los genes causantes conocidos y comprobar su utilidad diagnóstica en una cohorte de pacientes españoles.
MétodosEstudio transversal multicéntrico de pacientes con sospecha elevada de DCP, aplicando los criterios de la European Respiratory Society. Diseño de un panel de genes para secuenciación masiva con la tecnología de captura SeqCap EZ technology, incluyendo 44 genes relacionados con la DCP.
ResultadosSe incluyó a 79 pacientes de los que 53 presentaron un diagnóstico de DCP confirmado o muy probable. La sensibilidad del panel de genes fue del 81,1% con una especificidad del 100%. Se encontraron variantes candidatas en alguno de los genes del panel en 43 de los pacientes con DCP, siendo 51,2% (22/43) homocigotos y 48,8% (21/43) heterocigotos compuestos. Los genes causales más frecuentes fueron DNAH5 y CCDC39. Encontramos 52 variantes distintas, 36 no descritas previamente en la literatura.
ConclusionesEl diseño y la implementación de un panel de genes a medida tiene un alto rendimiento diagnóstico genético de la DCP, lo que permite conocer mejor la afectación causal de estos pacientes y sentar las bases para futuros abordajes terapéuticos.
Primary ciliary dyskinesia (PCD) is characterized by an alteration in the ciliary structure causing difficulty in the clearance of respiratory secretions. Diagnosis is complex and based on a combination of techniques. The objective of this study was to design a gene panel including all known causative genes, and to corroborate their diagnostic utility in a cohort of Spanish patients.
MethodsThis was a multicenter cross-sectional study of patients with a high suspicion of PCD, according to European Respiratory Society criteria, designed around a gene panel for massive sequencing using SeqCap EZ capture technology that included 44 genes associated with PCD.
ResultsWe included 79 patients, 53 of whom had a diagnosis of confirmed or highly probable PCD. The sensitivity of the gene panel was 81.1%, with a specificity of 100%. Candidate variants were found in some of the genes of the panel in 43 patients with PCD, 51.2% (22/43) of whom were homozygotes and 48.8% (21/43) compound heterozygotes. The most common causative genes were DNAH5 and CCDC39. We found 52 different variants, 36 of which were not previously described in the literature.
ConclusionsThe design and implementation of a tailored gene panel produces a high yield in the genetic diagnosis of PCD. This panel provides a better understanding of the causative factors involved in these patients and lays down the groundwork for future therapeutic approaches.
La discinesia ciliar primaria (DCP) es una enfermedad rara (1/15.000 recién nacidos) caracterizada por una alteración en la estructura y función ciliar que impide el correcto aclaramiento de las secreciones respiratorias1,2. Sus manifestaciones clínicas incluyen tos productiva, rinitis crónica, otitis de repetición, bronquitis recurrentes, bronquiectasias3, infertilidad masculina, subfertilidad femenina y situs inversus (50%)1,2 o heterotaxia (6-12%)4.
Aunque los síntomas son característicos, algunos de ellos son similares a los de otras enfermedades respiratorias, de manera que, el diagnóstico de DCP es complicado de realizar y se basa en una combinación de diferentes pruebas. La European Respiratory Society (ERS)5 y la American Thoracic Society (ATS)6 han realizado recomendaciones diagnósticas con enfoques y algoritmos diferentes. Así, un valor disminuido de óxido nítrico nasal (NOn), se considera en las recomendaciones de la ERS una prueba de cribado, mientras que la ATS estima que puede ser diagnóstico, siempre que se mida con un aparato de quimioluminiscencia en pacientes de 5 años o más y se haya descartado la fibrosis quística6.
La videomicroscopia de alta velocidad (HSVM, high speed video-microscopy), que analiza la frecuencia y el patrón del batido ciliar, tiene una alta sensibilidad y especificidad para el diagnóstico, aunque su interpretación tiene un componente de subjetividad y se puede ver alterada por las infecciones respiratorias7. La ERS la considera una técnica cuyo resultado alterado es altamente sospechoso del diagnóstico de DCP5, pero la ATS no la incluye en su algoritmo más que como una prueba de ayuda6. El estudio de las proteínas ciliares por inmunofluorescencia es una técnica prometedora8,9, aunque no se incluye todavía en las recomendaciones diagnósticas5,6.
Actualmente, la presencia de alteraciones en el estudio con microscopía electrónica (ME) (defectos de brazos externos, defectos de brazos externos e internos, defectos de brazos internos con desorganización microtubular y ausencia del par central) y el hallazgo de variantes patogénicas en el estudio genético se consideran indicadores confirmatorios de DCP5,6. Mientras que la ME es una técnica compleja y da lugar a numerosos falsos positivos y negativos5, la realización de estudios genéticos mediante secuenciación masiva está permitiendo realizar nuevas aproximaciones con mayor rentabilidad diagnóstica.
La DCP es una enfermedad causada por variantes en distintos genes que codifican proteínas del axonema ciliar. La mayoría de los genes asociados a DCP son autosómicos con herencia recesiva, a excepción de PIH1D3, descrito recientemente y ligado al cromosoma X10, y 2genes que causan DCP sindrómica: RPGR, ligada al cromosoma X, cuyas mutaciones dan lugar a DCP y retinitis pigmentaria11, y OFD1 cuyas mutaciones causan DCP y discapacidad intelectual12. Actualmente, se han descrito algo más de 40 genes asociados a DCP que permiten definir el diagnóstico molecular de aproximadamente 70% de los pacientes13.
El objetivo de este estudio es el diseño de un panel de secuenciación masiva que incluya todos los genes causantes de DCP conocidos hasta el momento y comprobar su utilidad diagnóstica en una cohorte de pacientes con sospecha clínica de DCP.
MétodosPacientesSe ha realizado un estudio transversal multicéntrico de una cohorte de pacientes remitidos a los centros diagnósticos de DCP del Hospital Universitari Vall d’Hebron (Barcelona) y del grupo de DCP de Valencia para su valoración por tener una historia clínica indicativa de DCP.
El proyecto fue aprobado por el Comité de Ética de los hospitales participantes y se solicitó para su inclusión la autorización de los padres o tutores legales para los niños menores de 12 años, de los padres o tutores y del paciente cuando su edad era entre 12 y 18 años, y de los mayores de 18 años.
Se incluyó a pacientes procedentes del Hospital Vall d’Hebron (n = 41), grupo de DCP de Valencia (n = 14), Hospital Sant Joan de Déu (Esplugues, Barcelona) (n = 14), Hospital Miguel Servet (Zaragoza) (n = 4), Hospital del Mar (Barcelona) (n = 2), Hospital Parc Taulí (Sabadell, Barcelona) (n = 1), Hospital Germans Trias i Pujol (Badalona, Barcelona) (n = 1), Hospital Clínic (Barcelona) (n = 1) y Hospital Son Llàtzer (Palma de Mallorca) (n = 1).
Se siguieron las recomendaciones de la ERS5 para clasificar a los pacientes como DCP confirmada (historia indicativa, alteraciones diagnósticas en el estudio con ME) o muy probable (historia sugestiva, NOn bajo, alteraciones en la HSVM) y como DCP muy improbable, con base en la valoración de los datos clínicos y escala PICADAR14, NOn, HSVM o ME.
La determinación de NOn se realizó usando un analizador de óxido nítrico por quimioluminiscencia (CLD 88sp NO-analyser, ECO MEDICS AG, Duerten, Suiza). La frecuencia y el patrón de batido ciliar se analizaron con una cámara de grabación de alta velocidad (MotionPro® X4, IDT, CA, EE. UU.) acoplada a un microscopio óptico.
Algunos datos de los pacientes 14 y 15 (tabla 1S, material suplementario) se han publicado previamente9.
Secuenciación masiva y análisis de datosSe extrajo ADN de sangre periférica mediante extracción automática magnética (Chemagic, Perkin-Elmer, Waltham, MA, EE. UU.) o extracción manual utilizando el kit Quick-DNATM Midiprep Plus Kit (Zymo Research, Irvine, CA, EE. UU.). Se determinó la concentración de ADN con el reactivo Qubit dsDNA BR Assay Kit en el fluorímetro Qubit 2.0.
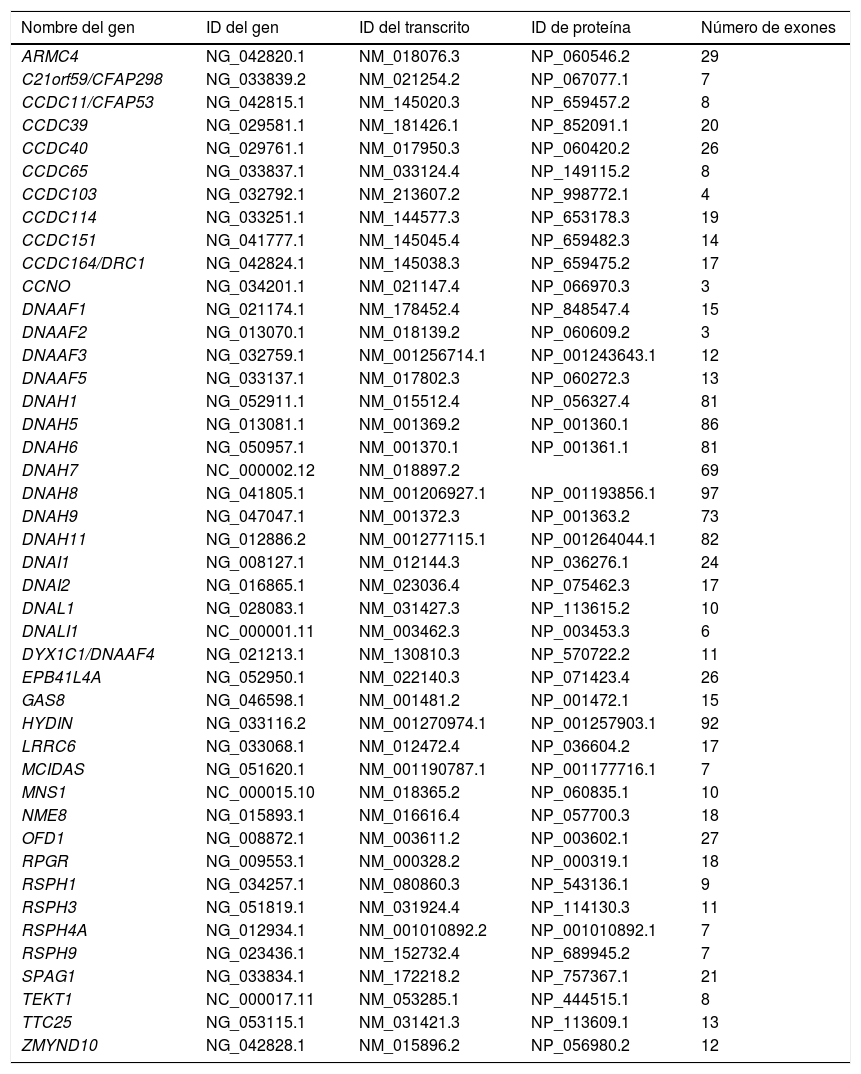
Para realizar el estudio genético se diseñó un panel que permite la secuenciación de exones y regiones intrónicas adyacentes (± 20 pb) a partir de la tecnología de captura SeqCap EZ (Roche Nimblegen, Pleasanton, CA, EE. UU.). En este panel se incluyeron 44 genes relacionados con la DCP, según lo descrito en la literatura en el momento del diseño (tabla 1).
Listado de genes incluidos en el panel para estudio de la discinesia ciliar primaria
| Nombre del gen | ID del gen | ID del transcrito | ID de proteína | Número de exones |
|---|---|---|---|---|
| ARMC4 | NG_042820.1 | NM_018076.3 | NP_060546.2 | 29 |
| C21orf59/CFAP298 | NG_033839.2 | NM_021254.2 | NP_067077.1 | 7 |
| CCDC11/CFAP53 | NG_042815.1 | NM_145020.3 | NP_659457.2 | 8 |
| CCDC39 | NG_029581.1 | NM_181426.1 | NP_852091.1 | 20 |
| CCDC40 | NG_029761.1 | NM_017950.3 | NP_060420.2 | 26 |
| CCDC65 | NG_033837.1 | NM_033124.4 | NP_149115.2 | 8 |
| CCDC103 | NG_032792.1 | NM_213607.2 | NP_998772.1 | 4 |
| CCDC114 | NG_033251.1 | NM_144577.3 | NP_653178.3 | 19 |
| CCDC151 | NG_041777.1 | NM_145045.4 | NP_659482.3 | 14 |
| CCDC164/DRC1 | NG_042824.1 | NM_145038.3 | NP_659475.2 | 17 |
| CCNO | NG_034201.1 | NM_021147.4 | NP_066970.3 | 3 |
| DNAAF1 | NG_021174.1 | NM_178452.4 | NP_848547.4 | 15 |
| DNAAF2 | NG_013070.1 | NM_018139.2 | NP_060609.2 | 3 |
| DNAAF3 | NG_032759.1 | NM_001256714.1 | NP_001243643.1 | 12 |
| DNAAF5 | NG_033137.1 | NM_017802.3 | NP_060272.3 | 13 |
| DNAH1 | NG_052911.1 | NM_015512.4 | NP_056327.4 | 81 |
| DNAH5 | NG_013081.1 | NM_001369.2 | NP_001360.1 | 86 |
| DNAH6 | NG_050957.1 | NM_001370.1 | NP_001361.1 | 81 |
| DNAH7 | NC_000002.12 | NM_018897.2 | 69 | |
| DNAH8 | NG_041805.1 | NM_001206927.1 | NP_001193856.1 | 97 |
| DNAH9 | NG_047047.1 | NM_001372.3 | NP_001363.2 | 73 |
| DNAH11 | NG_012886.2 | NM_001277115.1 | NP_001264044.1 | 82 |
| DNAI1 | NG_008127.1 | NM_012144.3 | NP_036276.1 | 24 |
| DNAI2 | NG_016865.1 | NM_023036.4 | NP_075462.3 | 17 |
| DNAL1 | NG_028083.1 | NM_031427.3 | NP_113615.2 | 10 |
| DNALI1 | NC_000001.11 | NM_003462.3 | NP_003453.3 | 6 |
| DYX1C1/DNAAF4 | NG_021213.1 | NM_130810.3 | NP_570722.2 | 11 |
| EPB41L4A | NG_052950.1 | NM_022140.3 | NP_071423.4 | 26 |
| GAS8 | NG_046598.1 | NM_001481.2 | NP_001472.1 | 15 |
| HYDIN | NG_033116.2 | NM_001270974.1 | NP_001257903.1 | 92 |
| LRRC6 | NG_033068.1 | NM_012472.4 | NP_036604.2 | 17 |
| MCIDAS | NG_051620.1 | NM_001190787.1 | NP_001177716.1 | 7 |
| MNS1 | NC_000015.10 | NM_018365.2 | NP_060835.1 | 10 |
| NME8 | NG_015893.1 | NM_016616.4 | NP_057700.3 | 18 |
| OFD1 | NG_008872.1 | NM_003611.2 | NP_003602.1 | 27 |
| RPGR | NG_009553.1 | NM_000328.2 | NP_000319.1 | 18 |
| RSPH1 | NG_034257.1 | NM_080860.3 | NP_543136.1 | 9 |
| RSPH3 | NG_051819.1 | NM_031924.4 | NP_114130.3 | 11 |
| RSPH4A | NG_012934.1 | NM_001010892.2 | NP_001010892.1 | 7 |
| RSPH9 | NG_023436.1 | NM_152732.4 | NP_689945.2 | 7 |
| SPAG1 | NG_033834.1 | NM_172218.2 | NP_757367.1 | 21 |
| TEKT1 | NC_000017.11 | NM_053285.1 | NP_444515.1 | 8 |
| TTC25 | NG_053115.1 | NM_031421.3 | NP_113609.1 | 13 |
| ZMYND10 | NG_042828.1 | NM_015896.2 | NP_056980.2 | 12 |
Los datos de ID se han obtenido de la base de datos National Center for Biotechnology Information (NCBI; https://www.ncbi.nlm.nih.gov/).
ID: identificación.
La captura de las regiones de interés se llevó a cabo de acuerdo con el protocolo comercial (SeqCap EZ [Roche Nimblegen, Pleasanton, CA, EE. UU.]), con una fragmentación enzimática de 21 min. La secuenciación de la librería se realizó mediante un secuenciador masivo de última generación MiSeq (Illumina, San Diego, CA, EE. UU.). El proceso de análisis de los datos incluyó el recorte de las secuencias con Trimmomantic (Institute for Biology, Aache, Alemania)15, el alineamiento de las secuencias con el genoma humano de referencia GRCh (hg38) usando BWA-MEM16, la detección de variantes con Genome Analysis Toolkit (GATK) Haplotype Caller (Broad Institute, Cambridge, MA, EE. UU.)17 y la anotación de las variantes con ANNOVAR18. Las variantes con una cobertura inferior a 20 no se tuvieron en cuenta en el análisis. La lista de las variantes identificadas se comparó con la información de bases de datos específicas para identificar variantes ya descritas en asociación a un fenotipo conocido (HGMD, ClinVar) y bases de datos de frecuencias poblacionales (GnomAD, ExAC, 1000 genomes) para descartar aquellas variantes que están presentes en la población general por encima del 1%. En paralelo, el análisis de datos también se realizó usando VariantStudio v2.2.1 (Illumina®, San Diego, CA, EE. UU.). Se evaluó la patogenicidad de las variantes utilizando el software Alamut v2.11 (Interactive Biosoftware, Ruan, Francia), que incluye Mutation Taster, Polyphen, Aling GVGD y SIFT, y Varsome (Saphetor, Lausana, Suiza), que incluye DANN, GERP y MutationTaster. Para mutaciones identificadas en regiones de splicing el efecto de las mismas se evaluó mediante SpliceSiteFinder, MaxEntScan, NNSPLICE, GeneSplicer y Human Splicing Finder, incluidos también en Alamut v2.11. Los datos de secuenciación masiva se reanalizaron bioinformáticamente utilizando el programa ExomeDepth19 para detectar variaciones en el número de copias (copy number variation [CNV]). La nomenclatura y la clasificación de las variantes está basada en las guías de la Human Genome Variation Society (HGVS) (https://www.hgvs.org/)20 y del American College of Medical Genetics and Genomics (ACMGG) (https://www.acmg.net/)21.
Las variantes probablemente patogénicas se confirmaron en los pacientes mediante secuenciación Sanger y, cuando fue posible, se analizó la cosegregación familiar.
Análisis estadísticoPara la descripción de las variables se han utilizado el porcentaje, la mediana y el rango y la media y desviación estándar (DE). Para el cálculo de la sensibilidad y especificidad del panel de genes, se consideraron como casos diagnosticados de DCP los casos de DCP confirmada o muy probable. Para la comparación entre los pacientes adultos y niños se ha utilizado la prueba de la chi al cuadrado, considerándose estadísticamente significativo un valor de p < 0,05. Los análisis se han realizado con el paquete estadístico MedCalc Statistical Software version 19.1.3 (MedCalc Software bvba, Ostend, Bélgica).
ResultadosEn el periodo comprendido entre enero del 2017 y noviembre del 2019 se estudió a 79 pacientes, pertenecientes a 74 familias distintas (74 casos índice), y 39 familiares. De los 79 pacientes, 26 se clasificaron como DCP muy improbable y en todos ellos el estudio genético fue negativo.
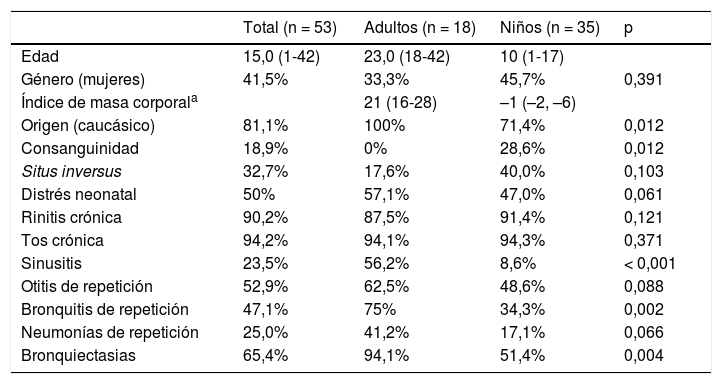
De los 53 pacientes con diagnóstico de DCP confirmado o muy probable, 35 eran pediátricos y 18 adultos. Cuarenta y tres pacientes eran de origen caucásico, 4 (7,5%) de origen marroquí, 4 (7,5%) paquistaní, uno de Oriente Medio y otro latinoamericano. Diez pacientes tenían una historia familiar de consanguinidad (tabla 2 y tabla 1S, material suplementario). Las manifestaciones clínicas más frecuentes fueron la tos crónica y la rinitis crónica. Un 50% tuvo antecedentes de distrés neonatal y el 32,7% presentaba situs inversus. La frecuencia de bronquiectasias fue superior en los pacientes adultos (94,1%) que en los pediátricos (51,4%) (tabla 2 y tabla 1S, material suplementario). El valor de la escala PICADAR fue igual o superior a 5 en 31 pacientes (65,9%). El NOn se pudo determinar en 35 casos, con un valor medio de 25,9 (DE 29,1) nl/min. En 25 el valor fue inferior a 33 nl/min y en solo 2 fue superior a 77 nl/min (tabla 1S, material suplementario). Los hallazgos de la HSVM y el ME se recogen en la tabla 1S, material suplementario. En 15 casos la alteración encontrada en el ME se consideró diagnóstica. La HSVM fue muy indicativa de DCP en 52 pacientes (no disponible en el paciente 3), siendo las alteraciones encontradas: patrón estático (n = 18), patrón estático con movimiento residual (n = 12), patrón rígido desorganizado (n = 8), patrón hipercinético (n = 5), patrón rotatorio (n = 6), discinesia (n = 2) y disminución movimiento distal (n = 1).
Características clínicas de los pacientes con discinesia ciliar primaria incluidos en el estudio
| Total (n = 53) | Adultos (n = 18) | Niños (n = 35) | p | |
|---|---|---|---|---|
| Edad | 15,0 (1-42) | 23,0 (18-42) | 10 (1-17) | |
| Género (mujeres) | 41,5% | 33,3% | 45,7% | 0,391 |
| Índice de masa corporala | 21 (16-28) | –1 (–2, –6) | ||
| Origen (caucásico) | 81,1% | 100% | 71,4% | 0,012 |
| Consanguinidad | 18,9% | 0% | 28,6% | 0,012 |
| Situs inversus | 32,7% | 17,6% | 40,0% | 0,103 |
| Distrés neonatal | 50% | 57,1% | 47,0% | 0,061 |
| Rinitis crónica | 90,2% | 87,5% | 91,4% | 0,121 |
| Tos crónica | 94,2% | 94,1% | 94,3% | 0,371 |
| Sinusitis | 23,5% | 56,2% | 8,6% | < 0,001 |
| Otitis de repetición | 52,9% | 62,5% | 48,6% | 0,088 |
| Bronquitis de repetición | 47,1% | 75% | 34,3% | 0,002 |
| Neumonías de repetición | 25,0% | 41,2% | 17,1% | 0,066 |
| Bronquiectasias | 65,4% | 94,1% | 51,4% | 0,004 |
Los datos se expresan como mediana y rango (entre paréntesis) para las variables cuantitativas (edad, índice de masa corporal) y como porcentaje para las variables cualitativas.
Las muestras de ADN se secuenciaron usando nuestro panel de genes, que ha cubierto el 98,75% de los exones y zonas intrónicas flanqueantes de los 44 genes incluidos (tabla 1). La cobertura media de los resultados fue de 600× con un 80,7% de reads on target.
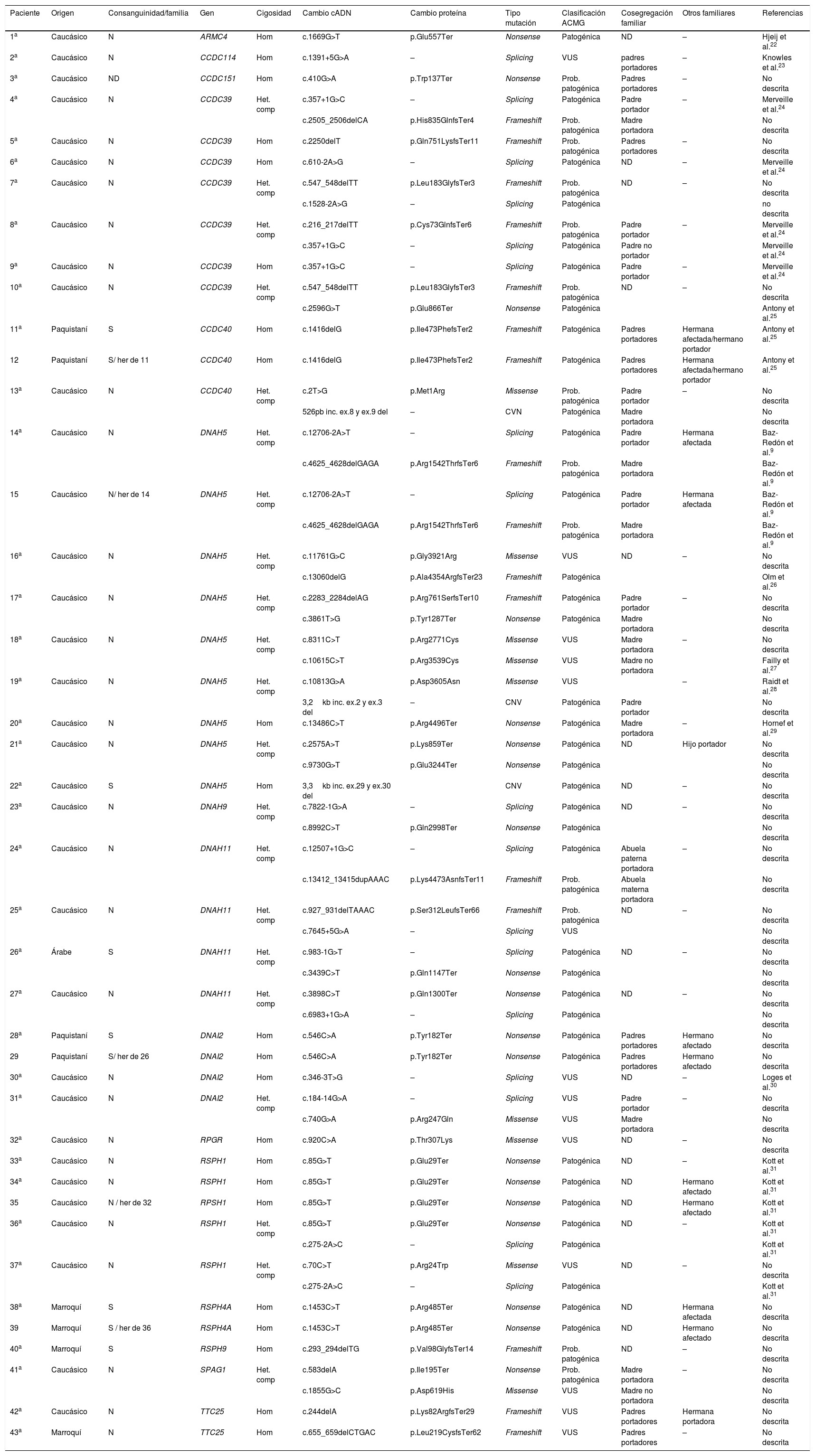
En 81,1% (43/53) de los pacientes con DCP se encontraron variantes candidatas en alguno de los genes del panel, siendo 22 (51,2%) homocigotos y 21 (48,8%) heterocigotos compuestos. En 18,9% (10/53) de los pacientes no se encontró ninguna variante que pudiera explicar el fenotipo (tabla 3). La sensibilidad de la técnica fue del 81,1% (IC del 95%, 68,0-90,6%) y la especificidad del 100% (IC del 95%, 86,8-100%). El área bajo la curva ROC fue de 0,91 (IC del 95%, 0,82-0,96). El valor predictivo positivo del panel de genes en nuestra población de estudio, en que la prevalencia de casos de DCP es del 67,1%, fue del 100% y el valor predictivo negativo del 72,2% (IC del 95%, 59,8-82,0%). Se han encontrado 52 variantes distintas (1 en ARMC4, 1 en CCDC114, 1 en CCDC151, 8 en CCDC39, 3 en CCDC40, 14 en DNAH5, 2 en DNAH9, 8 en DNAH11, 4 en DNAI2, 1 en RPGR, 3 en RSPH1, 1 en RSPH4A, 1 en RSPH9, 2 en SPAG1 y 2 en TTC25), 16 de las cuales habían sido previamente descritas asociadas a DCP9,22-31 y 36 no estaban descritas previamente en la literatura (tabla 3). De las 52 variantes encontradas, 14 (26,9%) nonsense, 13 (25%) fueron variantes frameshift, 13 (25%) de splicing, 9 (17,3%) missense y 3 (5,8%) CNV. Un 51,9% (27/52) se clasificaron como patogénicas (incluyendo las 3 CNV), un 21,2% (11/52) como probablemente patogénicas y un 26,9% (14/52) como variantes de significado incierto (VUS), según la clasificación del ACMG (tabla 3).
Resultados del estudio genético de los pacientes con variantes descritas que correlacionan con su fenotipo
| Paciente | Origen | Consanguinidad/familia | Gen | Cigosidad | Cambio cADN | Cambio proteína | Tipo mutación | Clasificación ACMG | Cosegregación familiar | Otros familiares | Referencias |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a | Caucásico | N | ARMC4 | Hom | c.1669G>T | p.Glu557Ter | Nonsense | Patogénica | ND | – | Hjeij et al.22 |
| 2a | Caucásico | N | CCDC114 | Hom | c.1391+5G>A | – | Splicing | VUS | padres portadores | – | Knowles et al.23 |
| 3a | Caucásico | ND | CCDC151 | Hom | c.410G>A | p.Trp137Ter | Nonsense | Prob. patogénica | Padres portadores | – | No descrita |
| 4a | Caucásico | N | CCDC39 | Het. comp | c.357+1G>C | – | Splicing | Patogénica | Padre portador | – | Merveille et al.24 |
| c.2505_2506delCA | p.His835GlnfsTer4 | Frameshift | Prob. patogénica | Madre portadora | No descrita | ||||||
| 5a | Caucásico | N | CCDC39 | Hom | c.2250delT | p.Gln751LysfsTer11 | Frameshift | Prob. patogénica | Padres portadores | – | No descrita |
| 6a | Caucásico | N | CCDC39 | Hom | c.610-2A>G | – | Splicing | Patogénica | ND | – | Merveille et al.24 |
| 7a | Caucásico | N | CCDC39 | Het. comp | c.547_548delTT | p.Leu183GlyfsTer3 | Frameshift | Prob. patogénica | ND | – | No descrita |
| c.1528-2A>G | – | Splicing | Patogénica | no descrita | |||||||
| 8a | Caucásico | N | CCDC39 | Het. comp | c.216_217delTT | p.Cys73GlnfsTer6 | Frameshift | Prob. patogénica | Padre portador | – | Merveille et al.24 |
| c.357+1G>C | – | Splicing | Patogénica | Padre no portador | Merveille et al.24 | ||||||
| 9a | Caucásico | N | CCDC39 | Hom | c.357+1G>C | – | Splicing | Patogénica | Padre portador | – | Merveille et al.24 |
| 10a | Caucásico | N | CCDC39 | Het. comp | c.547_548delTT | p.Leu183GlyfsTer3 | Frameshift | Prob. patogénica | ND | – | No descrita |
| c.2596G>T | p.Glu866Ter | Nonsense | Patogénica | Antony et al.25 | |||||||
| 11a | Paquistaní | S | CCDC40 | Hom | c.1416delG | p.Ile473PhefsTer2 | Frameshift | Patogénica | Padres portadores | Hermana afectada/hermano portador | Antony et al.25 |
| 12 | Paquistaní | S/ her de 11 | CCDC40 | Hom | c.1416delG | p.Ile473PhefsTer2 | Frameshift | Patogénica | Padres portadores | Hermana afectada/hermano portador | Antony et al.25 |
| 13a | Caucásico | N | CCDC40 | Het. comp | c.2T>G | p.Met1Arg | Missense | Prob. patogénica | Padre portador | – | No descrita |
| 526pb inc. ex.8 y ex.9 del | – | CVN | Patogénica | Madre portadora | No descrita | ||||||
| 14a | Caucásico | N | DNAH5 | Het. comp | c.12706-2A>T | – | Splicing | Patogénica | Padre portador | Hermana afectada | Baz-Redón et al.9 |
| c.4625_4628delGAGA | p.Arg1542ThrfsTer6 | Frameshift | Prob. patogénica | Madre portadora | Baz-Redón et al.9 | ||||||
| 15 | Caucásico | N/ her de 14 | DNAH5 | Het. comp | c.12706-2A>T | – | Splicing | Patogénica | Padre portador | Hermana afectada | Baz-Redón et al.9 |
| c.4625_4628delGAGA | p.Arg1542ThrfsTer6 | Frameshift | Prob. patogénica | Madre portadora | Baz-Redón et al.9 | ||||||
| 16a | Caucásico | N | DNAH5 | Het. comp | c.11761G>C | p.Gly3921Arg | Missense | VUS | ND | – | No descrita |
| c.13060delG | p.Ala4354ArgfsTer23 | Frameshift | Patogénica | Olm et al.26 | |||||||
| 17a | Caucásico | N | DNAH5 | Het. comp | c.2283_2284delAG | p.Arg761SerfsTer10 | Frameshift | Patogénica | Padre portador | – | No descrita |
| c.3861T>G | p.Tyr1287Ter | Nonsense | Patogénica | Madre portadora | No descrita | ||||||
| 18a | Caucásico | N | DNAH5 | Het. comp | c.8311C>T | p.Arg2771Cys | Missense | VUS | Madre portadora | – | No descrita |
| c.10615C>T | p.Arg3539Cys | Missense | VUS | Madre no portadora | Failly et al.27 | ||||||
| 19a | Caucásico | N | DNAH5 | Het. comp | c.10813G>A | p.Asp3605Asn | Missense | VUS | – | Raidt et al.28 | |
| 3,2kb inc. ex.2 y ex.3 del | – | CNV | Patogénica | Padre portador | No descrita | ||||||
| 20a | Caucásico | N | DNAH5 | Hom | c.13486C>T | p.Arg4496Ter | Nonsense | Patogénica | Madre portadora | – | Hornef et al.29 |
| 21a | Caucásico | N | DNAH5 | Het. comp | c.2575A>T | p.Lys859Ter | Nonsense | Patogénica | ND | Hijo portador | No descrita |
| c.9730G>T | p.Glu3244Ter | Nonsense | Patogénica | No descrita | |||||||
| 22a | Caucásico | S | DNAH5 | Hom | 3,3kb inc. ex.29 y ex.30 del | CNV | Patogénica | ND | – | No descrita | |
| 23a | Caucásico | N | DNAH9 | Het. comp | c.7822-1G>A | – | Splicing | Patogénica | ND | – | No descrita |
| c.8992C>T | p.Gln2998Ter | Nonsense | Patogénica | No descrita | |||||||
| 24a | Caucásico | N | DNAH11 | Het. comp | c.12507+1G>C | – | Splicing | Patogénica | Abuela paterna portadora | – | No descrita |
| c.13412_13415dupAAAC | p.Lys4473AsnfsTer11 | Frameshift | Prob. patogénica | Abuela materna portadora | No descrita | ||||||
| 25a | Caucásico | N | DNAH11 | Het. comp | c.927_931delTAAAC | p.Ser312LeufsTer66 | Frameshift | Prob. patogénica | ND | – | No descrita |
| c.7645+5G>A | – | Splicing | VUS | No descrita | |||||||
| 26a | Árabe | S | DNAH11 | Het. comp | c.983-1G>T | – | Splicing | Patogénica | ND | – | No descrita |
| c.3439C>T | p.Gln1147Ter | Nonsense | Patogénica | No descrita | |||||||
| 27a | Caucásico | N | DNAH11 | Het. comp | c.3898C>T | p.Gln1300Ter | Nonsense | Patogénica | ND | – | No descrita |
| c.6983+1G>A | – | Splicing | Patogénica | No descrita | |||||||
| 28a | Paquistaní | S | DNAI2 | Hom | c.546C>A | p.Tyr182Ter | Nonsense | Patogénica | Padres portadores | Hermano afectado | No descrita |
| 29 | Paquistaní | S/ her de 26 | DNAI2 | Hom | c.546C>A | p.Tyr182Ter | Nonsense | Patogénica | Padres portadores | Hermano afectado | No descrita |
| 30a | Caucásico | N | DNAI2 | Hom | c.346-3T>G | – | Splicing | VUS | ND | – | Loges et al.30 |
| 31a | Caucásico | N | DNAI2 | Het. comp | c.184-14G>A | – | Splicing | VUS | Padre portador | – | No descrita |
| c.740G>A | p.Arg247Gln | Missense | VUS | Madre portadora | No descrita | ||||||
| 32a | Caucásico | N | RPGR | Hom | c.920C>A | p.Thr307Lys | Missense | VUS | ND | – | No descrita |
| 33a | Caucásico | N | RSPH1 | Hom | c.85G>T | p.Glu29Ter | Nonsense | Patogénica | ND | – | Kott et al.31 |
| 34a | Caucásico | N | RSPH1 | Hom | c.85G>T | p.Glu29Ter | Nonsense | Patogénica | ND | Hermano afectado | Kott et al.31 |
| 35 | Caucásico | N / her de 32 | RPSH1 | Hom | c.85G>T | p.Glu29Ter | Nonsense | Patogénica | ND | Hermano afectado | Kott et al.31 |
| 36a | Caucásico | N | RSPH1 | Het. comp | c.85G>T | p.Glu29Ter | Nonsense | Patogénica | ND | – | Kott et al.31 |
| c.275-2A>C | – | Splicing | Patogénica | Kott et al.31 | |||||||
| 37a | Caucásico | N | RSPH1 | Het. comp | c.70C>T | p.Arg24Trp | Missense | VUS | ND | – | No descrita |
| c.275-2A>C | – | Splicing | Patogénica | Kott et al.31 | |||||||
| 38a | Marroquí | S | RSPH4A | Hom | c.1453C>T | p.Arg485Ter | Nonsense | Patogénica | ND | Hermana afectada | No descrita |
| 39 | Marroquí | S / her de 36 | RSPH4A | Hom | c.1453C>T | p.Arg485Ter | Nonsense | Patogénica | ND | Hermano afectado | No descrita |
| 40a | Marroquí | S | RSPH9 | Hom | c.293_294delTG | p.Val98GlyfsTer14 | Frameshift | Prob. patogénica | ND | – | No descrita |
| 41a | Caucásico | N | SPAG1 | Het. comp | c.583delA | p.Ile195Ter | Nonsense | Prob. patogénica | Madre portadora | – | No descrita |
| c.1855G>C | p.Asp619His | Missense | VUS | Madre no portadora | No descrita | ||||||
| 42a | Caucásico | N | TTC25 | Hom | c.244delA | p.Lys82ArgfsTer29 | Frameshift | VUS | Padres portadores | Hermana portadora | No descrita |
| 43a | Marroquí | N | TTC25 | Hom | c.655_659delCTGAC | p.Leu219CysfsTer62 | Frameshift | VUS | Padres portadores | – | No descrita |
ACMG: American College of Medical Genetics; ex.: exón; Her: hermano; Het. comp: heterocigoto compuesto; Het: heterocigoto; Hom: homocigoto; kb: kilobases; ND: muestra no disponible; pb: pares de bases; Prob. patogénica: probablemente patogénica; VUS: variante de significado incierto; –: falta de datos.
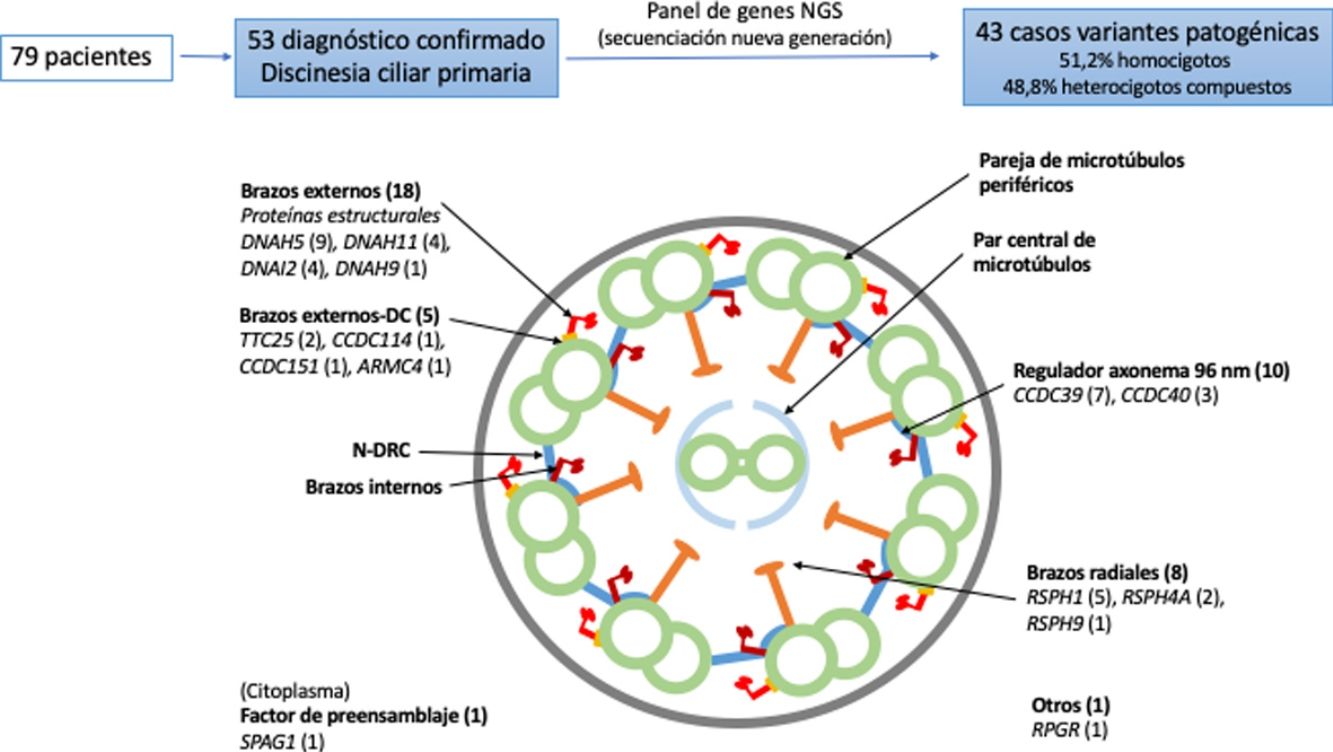
Dieciocho pacientes presentaron variantes en genes relacionados con proteínas estructurales de los brazos externos de dineína (DNAH5 [n = 9], DNAH11 [n = 4], DNAI2 [n = 4], DNAH9 [n = 1]) y 5 con el docking complex de los brazos externos de dineína (TTC25 [n = 2], ARMC4 [n = 1], CCDC114 [n = 1], CCDC151 [n = 1]); 8 mostraron variantes en genes que codifican proteínas de los brazos radiales (RSPH1 [n = 5], RSPH4A [n = 2], RSPH9 [n = 1]); en 10 se detectaron variantes en genes que codifican proteínas del complejo regulador del axonema (CCDC39 [n = 7], CCDC40 (n = 3]); un paciente presentó variantes en SPAG1, que codifica una proteína probablemente relacionada con el transporte o ensamblaje citoplasmático de los complexos de dineína, y uno en RPGR, gen asociado a retinitis pigmentaria (fig. 1, tabla 3). Las variantes en los 3genes más frecuentes (DNAH5, CCDC39 y RSPH1) se presentaron solo en los pacientes de origen caucásico. En los pacientes de origen no caucásico, los genes causales más frecuentes fueron CCDC40, DNAI2 y RSPH4A, con 2 casos cada uno.

Diagrama transversal de un cilio indicando sus componentes estructurales y los genes en los que se han encontrado variantes. Se indica entre paréntesis el número de pacientes con variantes en cada gen. Brazos externos-DC: docking complex de los brazos externos de dineína; N-DRC: nexina-complexo regulador de la dineína.
Se ha analizado con el panel de genes a 37 familiares de 22 familias distintas. Todos los padres analizados (18 familias distintas) eran portadores de alguna de las variantes encontradas en sus hijos. Se analizaron los ADN procedentes de los abuelos maternos y la abuela materna del paciente 24 y se determinó que la variante c.12507+1G>C es de origen paterno y la c.13412_13415dupAAAC de origen materno (tabla 3).
DiscusiónEn una cohorte de 53 pacientes con DCP confirmada o muy probable, utilizando un panel de 44 genes mediante secuencia masiva, se obtuvieron resultados genéticos positivos en el 81,1% de los pacientes, pudiendo describir, por lo tanto, en ellos el gen causante del defecto de la estructura ciliar. En otros 26 pacientes, remitidos por clínica respiratoria sospechosa, pero con diagnóstico de DCP muy improbable tras realizar las pruebas previas, el estudio genético fue negativo. Este es el primer estudio, en nuestro conocimiento, en describir los genes causantes de discinesia ciliar en una cohorte amplia de pacientes en España.
Con nuestros resultados hemos confirmado que, aplicado a nuestra población, el panel de genes tiene un alto rendimiento diagnóstico (sensibilidad del 81,1%) y que pudo descartar a todos los pacientes con baja sospecha de DCP (especificidad del 100%). El rendimiento de los paneles de genes aplicados a otras poblaciones ha ido aumentado a medida que se van descubriendo nuevos genes y se van incorporando a los paneles, variando entre un 43 y un 70%32-34, y más recientemente un 82%35.
El diagnóstico de la DCP es complejo con las técnicas disponibles hasta ahora, lo que genera en los médicos y pacientes muchas incertezas diagnósticas y dudas acerca del pronóstico y evolución de su enfermedad. La determinación de NOn con punto de corte 77 nl/min tiene una alta sensibilidad (93,6%), pero una especificidad del 78,9%36. El microscopio electrónico es específico (100%), pero no identifica un 21% de los casos, necesita personas muy expertas para su interpretación y a veces no se consigue una muestra adecuada5,37. La HSVM tiene una excelente sensibilidad y especificidad, pero también necesita un equipo experto y a menudo repetir la prueba varias veces8. El estudio genético, aunque también puede no identificar un 20% de los casos, permite obtener un diagnóstico de certeza, lo que ayuda a orientar de forma más adecuada el tratamiento de los pacientes y a poder realizar un consejo genético y sentar las bases para la investigación de tratamientos específicos, como podrían ser la terapia génica o terapias proteicas13.
La secuenciación masiva con el panel de genes permite el estudio de variantes puntuales y de deleciones o inserciones pequeñas (indels) y las variantes en número de copia (CNV) de los genes descritos hasta el momento como causantes de DCP. Con esta técnica, una proporción de pacientes con DCP confirmada o muy probable, 18,9% en nuestra serie, queda sin diagnóstico genético. En ellos, el análisis del exoma completo podría ayudar a encontrar nuevos genes causantes de DCP.
La mayoría de las variantes descritas en nuestros pacientes (82,7%) causaron la pérdida de función proteica (nonsense, frameshift, CNV y splicing), resultados similares a los descritos en otros estudios35. En 9 (17,3%) encontramos variantes missense de cambio de un único aminoácido y se catalogaron como VUS según la clasificación ACMG21, a excepción de la variante c.2T>G/p.Met1Arg (paciente 13) que afectaba al primer aminoácido y se clasificó como probablemente patogénica (tabla 3). Estas variantes missense se tuvieron en cuenta como causa posible de alteración proteica según las predicciones in silico. Idealmente, estos defectos missense se deberían comprobar in vitro mediante cultivos de células de epitelio respiratorio nasal del paciente o modelos animales.
Dado el elevado número de variantes que se pueden encontrar con la secuenciación masiva, muchas de ellas benignas, es fundamental para interpretar de forma correcta los resultados relacionar las variantes encontradas con los hallazgos del ME y la HSVM. En nuestra serie, hubo una buena relación entre la ultraestructura y los hallazgos genéticos en solo 6 casos, dadas las dificultades de interpretación de la ME y las posibilidades de alteraciones por infecciones respiratorias o por artefactos de procesamiento9,37, mientras que el estudio con HSVM mostró una buena relación con los hallazgos genéticos en todos los casos.
Los pacientes 13 y 19, que tenían inicialmente una única variante en heterocigosis en los genes CCDC40 y DNAH5, respectivamente, se pudieron resolver a partir del análisis bioinformático de CNV, así como la paciente 22. En los pacientes 13 y 19 se describieron deleciones en el otro alelo que concordaba, además, con el estudio de segregación familiar, siendo la madre de la paciente 13 y el padre del paciente 19 los portadores de estas deleciones (tabla 3). En la paciente 22 se ha detectado una deleción en homocigosis en el gen DNAH5. El análisis bioinformático de CNV es una herramienta útil para resolver algunos casos, sobre todo en aquellos con variantes monoalélicas en un gen candidato que cuadre con el fenotipo.
Cabe destacar que todos los casos de consanguinidad de nuestra cohorte tuvieron resultado molecular positivo y fueron homocigotos para las variantes encontradas, todas ellas clasificadas como patogénicas o probablemente patogénicas (tabla 3).
La distribución de genes causantes de DCP puede ser diferente dependiendo del origen étnico35. En nuestra serie DNAH5 y CCDC39 han sido los genes de mayor incidencia y los 2se encontraron solo en los pacientes de origen caucásico (tabla 3). DNAH5 se ha descrito como el gen más frecuente en estudios en población caucásica, explicando el 15-37% de los casos27,32,33,35 y poco frecuente en otras poblaciones como la árabe34,35. El gen CCDC39 se ha descrito previamente en pacientes de origen europeo25 y es uno de los genes que predomina en población de origen árabe34,35.
Las limitaciones de nuestro estudio están relacionadas principalmente con el número de pacientes estudiados que, aunque significativo para una enfermedad rara, hay que ampliar para conocer mejor la frecuencia de las diferentes variantes en nuestra población, tanto en los de origen caucásico, como no caucásico. La secuenciación masiva no permite detectar todas las deleciones/duplicaciones en los genes, lo que se ha solventado a través del análisis bioinformático con el análisis de CNV, aunque este es solo una aproximación y es conveniente confirmarlas con otros métodos. Otras limitaciones son las inherentes al estudio con un panel de genes, ya que no se analiza todo el exoma ni todo el genoma. Sin embargo, ello facilita la interpretación de los resultados, ya que en el análisis del exoma completo o del genoma pueden aparecer un número muy elevado de variantes sin significado patogénico en población sana. Además, en los paneles de genes, la cobertura de estos está optimizada respecto al estudio del exoma. Aunque el panel de genes diseñado a medida ha permitido conocer el defecto específico de los pacientes diagnosticados molecularmente, dado que cada año se describen nuevos genes de DCP38-40, es necesario una ampliación de este panel con los genes descubiertos hasta la fecha.
En conclusión, los resultados de este estudio muestran la utilidad del diseño y la implementación del análisis genético mediante paneles de genes a medida, que representa una herramienta de utilidad para conseguir un mejor diagnóstico de la DCP.
FinanciaciónEl presente trabajo ha sido financiado por una ayuda de Acción Estratégica en Salud del Instituto de Salud Carlos III (ISCIII) (PI16/01233), cofinanciadas por el Fondo Europeo de Desarrollo Regional, Programa Operativo Crecimiento Inteligente 2014-2020, una beca de la Sociedad Española de Neumología (2016) Pediátrica y una beca de la Fundació Catalana de Pneumologia (FUCAP) (2016). NCT recibió una ayuda para una Short term Scientific Mission de la COST Action BM1407.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Los autores participan en la COST Action BM1407 Translational research in primary ciliary dyskinesia: bench, bedside, and population perspectives (BEAT PCD). AMG y SRA participan en la ERN-LUNG. Este trabajo se ha realizado en el marco del programa de doctorado de Pediatría, Obstetricia y Ginecología de la Universitat Autònoma de Barcelona. Damos la gracias al Dr. Josep Quer y a Damir García-Cehic (Vall d’Hebron Institut de Recerca [VHIR], Barcelona) por su valiosa colaboración y por su excelente asistencia en las técnicas de secuenciación masiva.




www.publicationethics.org.




Archivos de Bronconeumología follows the Recommendations for the Conduct, Reporting, Editing and Publication of Scholarly Work in Medical Journals