

Primary ciliary dyskinesia (PCD) is characterized by an alteration in the ciliary structure causing difficulty in the clearance of respiratory secretions. Diagnosis is complex and based on a combination of techniques. The objective of this study was to design a gene panel including all known causative genes, and to corroborate their diagnostic utility in a cohort of Spanish patients.
MethodsThis was a multicenter cross-sectional study of patients with a high suspicion of PCD according to European Respiratory Society criteria. We designed a gene panel for massive sequencing using SeqCap EZ capture technology that included 44 genes associated with PCD.
ResultsWe included 79 patients, 53 of whom had a diagnosis of confirmed or highly probable PCD. The sensitivity of the gene panel was 81.1%, with a specificity of 100%. Candidate variants were found in some of the genes of the panel in 43 patients with PCD, 51.2% (22/43) of whom were homozygotes and 48.8% (21/43) compound heterozygotes. The most common causative genes were DNAH5 and CCDC39. We found 52 different variants, 36 of which were not previously described in the literature.
ConclusionsThe design and implementation of a tailored gene panel produces a high yield in the genetic diagnosis of PCD. This panel provides a better understanding of the causative factors involved in these patients and lays down the groundwork for future therapeutic approaches.
La discinesia ciliar primaria (DCP) es una enfermedad caracterizada por una alteración en la estructura ciliar que impide el correcto aclaramiento de las secreciones respiratorias. Su diagnóstico es complejo y se basa en una combinación de técnicas. El objetivo de este estudio fue diseñar un panel de genes incluyendo todos los genes causantes conocidos y comprobar su utilidad diagnóstica en una cohorte de pacientes españoles.
MétodosEstudio transversal multicéntrico de pacientes con sospecha elevada de DCP, aplicando los criterios de la European Respiratory Society. Diseño de un panel de genes para secuenciación masiva con la tecnología de captura SeqCap EZ technology, incluyendo 44 genes relacionados con la DCP.
ResultadosSe incluyó a 79 pacientes de los que 53 presentaron un diagnóstico de DCP confirmado o muy probable. La sensibilidad del panel de genes fue del 81,1% con una especificidad del 100%. Se encontraron variantes candidatas en alguno de los genes del panel en 43 de los pacientes con DCP, siendo 51,2% (22/43) homocigotos y 48,8% (21/43) heterocigotos compuestos. Los genes causales más frecuentes fueron DNAH5 y CCDC39. Encontramos 52 variantes distintas, 36 no descritas previamente en la literatura.
ConclusionesEl diseño y la implementación de un panel de genes a medida tiene un alto rendimiento diagnóstico genético de la DCP, lo que permite conocer mejor la afectación causal de estos pacientes y sentar las bases para futuros abordajes terapéuticos.
Primary ciliary dyskinesia (PCD) is a rare disease, occurring in 1/15,000 newborns. It is characterized by an alteration in ciliary structure and function that prevents the correct clearance of respiratory secretions.1,2 Clinical manifestations include productive cough, chronic rhinitis, recurrent otitis, recurrent bronchitis, bronchiectasis,3 male infertility, female subfertility, situs inversus (50%),1,2 and heterotaxy (6-12%).4
It presents with characteristic symptoms, but some are similar to those of other respiratory diseases: PCD is therefore difficult to diagnose and the process is based on a combination of different tests. The European Respiratory Society (ERS)5 and the American Thoracic Society (ATS)6 have made diagnostic recommendations using different approaches and algorithms. In the ERS recommendations, for example, low nasal nitric oxide (nNO) is considered a screening test, while according to the ATS this result can be diagnostic if it is measured with a chemiluminescence analyzer in patients at least 5 years of age, after ruling out cystic fibrosis.6
High-speed videomicroscopy (HSVM), which analyzes ciliary beat pattern and beat frequency, is highly sensitive and specific for diagnosis, although its interpretation has a subjective component and results can be altered by respiratory infections.7 In the opinion of the ERS, an anomalous result on this test is highly suggestive of a diagnosis of PCD,5 but the ATS does not include it in its algorithm other than as supplementary test.6 Immunofluorescence study of ciliary proteins is a promising technique,8,9 although it has not yet been included in the diagnostic recommendations.5,6
Currently, the presence of alterations on electron microscopy (EM) (outer dynein arm defects, inner and outer dynein arm defects, inner dynein arm defects with microtubular disorganization, and central pair absence) and the finding of pathogenic variants in the genetic study are considered indicators that confirm PCD.5,6 While EM is a complex technique that gives numerous false positives and negatives,5 genetic studies using massive sequencing technology are opening up new approaches that offer greater diagnostic yield.
PCD is a disease caused by variants in different genes encoding ciliary axoneme proteins. Most genes associated with PCD are autosomal recessive, with the exception of the recently described PIH1D3 that is linked to the X chromosome,10 and 2 genes that cause syndromic PCD: RPGR, linked to the X chromosome, whose mutations give rise to PCD and retinitis pigmentosa,11 and OFD1, whose mutations cause PCD and intellectual impairment.12 At present, just over 40 genes associated with PCD that define the molecular diagnosis of approximately 70% of patients have been described.13
The aim of this study was to design a massive sequencing panel that includes all known genes causing PCD and to verify their diagnostic usefulness in a cohort of patients with clinical suspicion of PCD.
MethodsPatientsWe performed a multicenter, cross-sectional study of a cohort of patients with a clinical history indicative of PCD referred for assessment to the PCD diagnostic center of the Hospital Universitari Vall d’Hebron (Barcelona) and the PCD group in Valencia.
The project was approved by the Ethics Committee of the participating hospitals and authorization for inclusion was requested from parents or legal guardians of children under the age of 12; from the parents or guardians and patients aged between 12 and 18; and from patients over 18 years of age.
Patients were included from the Vall d’Hebron Hospital (n = 41), the PCD group in Valencia (n = 14), Hospital Sant Joan de Déu (Esplugues, Barcelona) (n = 14), Hospital Miguel Servet (Zaragoza) (n = 4), Hospital del Mar (Barcelona) (n = 2), Hospital Parc Taulí (Sabadell, Barcelona) (n = 1), Germans Trias i Pujol Hospital (Badalona, Barcelona) (n = 1), Hospital Clínic (Barcelona) (n = 1), and Hospital Son Llàtzer (Palma de Mallorca) (n = 1).
ERS recommendations5 were followed to classify patients as confirmed (indicative history, diagnostic alterations on EM) or very likely PCD (suggestive history, low nNO, changes on HSVM), or as highly unlikely PCD, based on the evaluation of clinical data and the PICADAR score,14 nNO, HSVM or EM.
A chemiluminescent nitric oxide analyzer (CLD 88sp NO-analysis, ECO MEDICS AG, Duerten, Switzerland) was used to determine nNO. Ciliary beat pattern and frequency were analyzed with a high-speed digital camera (MotionPro® X4, IDT, CA, USA) connected to an optical microscope.
Some data from patients 14 and 15 (Appendix B Table 1S, supplemental material) have been previously published.9
Massive sequencing and data analysisDNA was extracted from peripheral blood by magnetic extraction (Chemagic, Perkin-Elmer, Waltham, MA, USA) or by manual extraction using the Quick-DNA™ Midiprep Plus Kit (Zymo Research, Irvine, CA, USA). DNA concentration was determined with the Qubit dsDNA BR Assay Kit reagent on the Qubit 2.0 fluorometer.
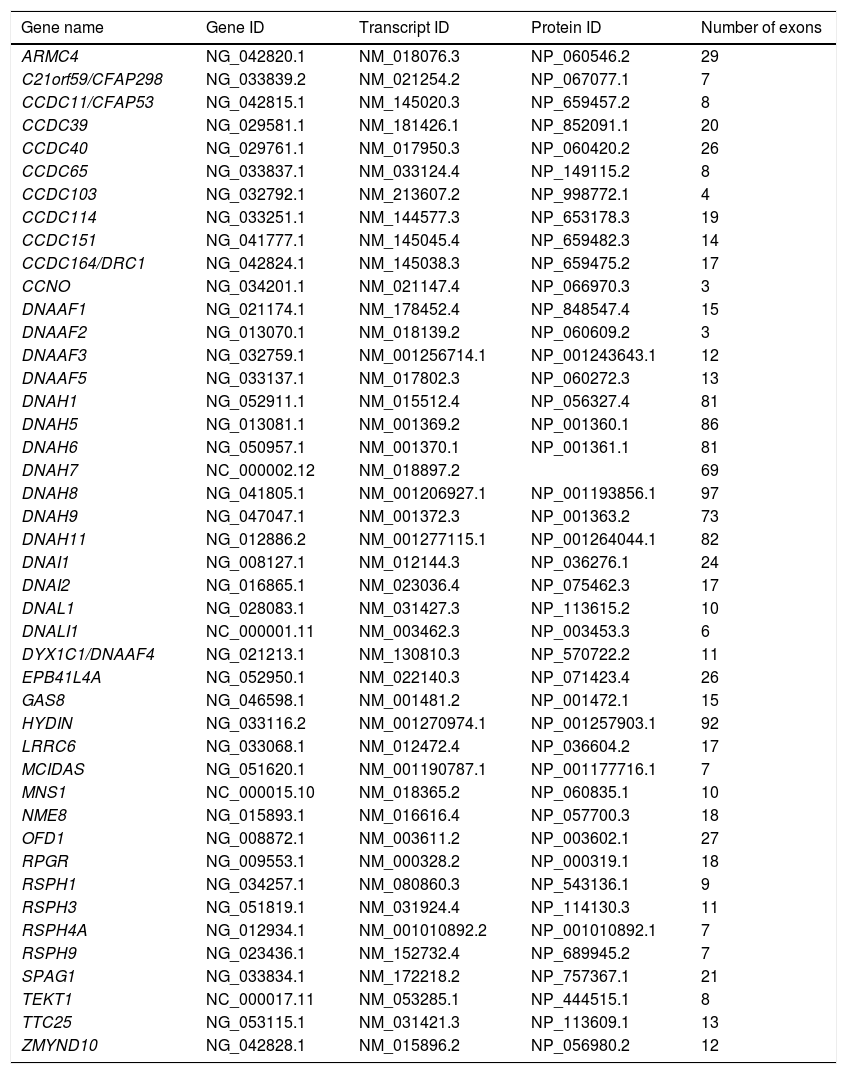
For the genetic study, a panel was designed for the sequencing of exons and their flanking intronic regions (± 20 bp) using SeqCap EZ capture technology (Roche NimbleGen, Pleasanton, CA, USA). This panel included 44 genes related to PCD, described in the literature at the time of design (Table 1).
List of genes included in the primary ciliary dyskinesia panel.
| Gene name | Gene ID | Transcript ID | Protein ID | Number of exons |
|---|---|---|---|---|
| ARMC4 | NG_042820.1 | NM_018076.3 | NP_060546.2 | 29 |
| C21orf59/CFAP298 | NG_033839.2 | NM_021254.2 | NP_067077.1 | 7 |
| CCDC11/CFAP53 | NG_042815.1 | NM_145020.3 | NP_659457.2 | 8 |
| CCDC39 | NG_029581.1 | NM_181426.1 | NP_852091.1 | 20 |
| CCDC40 | NG_029761.1 | NM_017950.3 | NP_060420.2 | 26 |
| CCDC65 | NG_033837.1 | NM_033124.4 | NP_149115.2 | 8 |
| CCDC103 | NG_032792.1 | NM_213607.2 | NP_998772.1 | 4 |
| CCDC114 | NG_033251.1 | NM_144577.3 | NP_653178.3 | 19 |
| CCDC151 | NG_041777.1 | NM_145045.4 | NP_659482.3 | 14 |
| CCDC164/DRC1 | NG_042824.1 | NM_145038.3 | NP_659475.2 | 17 |
| CCNO | NG_034201.1 | NM_021147.4 | NP_066970.3 | 3 |
| DNAAF1 | NG_021174.1 | NM_178452.4 | NP_848547.4 | 15 |
| DNAAF2 | NG_013070.1 | NM_018139.2 | NP_060609.2 | 3 |
| DNAAF3 | NG_032759.1 | NM_001256714.1 | NP_001243643.1 | 12 |
| DNAAF5 | NG_033137.1 | NM_017802.3 | NP_060272.3 | 13 |
| DNAH1 | NG_052911.1 | NM_015512.4 | NP_056327.4 | 81 |
| DNAH5 | NG_013081.1 | NM_001369.2 | NP_001360.1 | 86 |
| DNAH6 | NG_050957.1 | NM_001370.1 | NP_001361.1 | 81 |
| DNAH7 | NC_000002.12 | NM_018897.2 | 69 | |
| DNAH8 | NG_041805.1 | NM_001206927.1 | NP_001193856.1 | 97 |
| DNAH9 | NG_047047.1 | NM_001372.3 | NP_001363.2 | 73 |
| DNAH11 | NG_012886.2 | NM_001277115.1 | NP_001264044.1 | 82 |
| DNAI1 | NG_008127.1 | NM_012144.3 | NP_036276.1 | 24 |
| DNAI2 | NG_016865.1 | NM_023036.4 | NP_075462.3 | 17 |
| DNAL1 | NG_028083.1 | NM_031427.3 | NP_113615.2 | 10 |
| DNALI1 | NC_000001.11 | NM_003462.3 | NP_003453.3 | 6 |
| DYX1C1/DNAAF4 | NG_021213.1 | NM_130810.3 | NP_570722.2 | 11 |
| EPB41L4A | NG_052950.1 | NM_022140.3 | NP_071423.4 | 26 |
| GAS8 | NG_046598.1 | NM_001481.2 | NP_001472.1 | 15 |
| HYDIN | NG_033116.2 | NM_001270974.1 | NP_001257903.1 | 92 |
| LRRC6 | NG_033068.1 | NM_012472.4 | NP_036604.2 | 17 |
| MCIDAS | NG_051620.1 | NM_001190787.1 | NP_001177716.1 | 7 |
| MNS1 | NC_000015.10 | NM_018365.2 | NP_060835.1 | 10 |
| NME8 | NG_015893.1 | NM_016616.4 | NP_057700.3 | 18 |
| OFD1 | NG_008872.1 | NM_003611.2 | NP_003602.1 | 27 |
| RPGR | NG_009553.1 | NM_000328.2 | NP_000319.1 | 18 |
| RSPH1 | NG_034257.1 | NM_080860.3 | NP_543136.1 | 9 |
| RSPH3 | NG_051819.1 | NM_031924.4 | NP_114130.3 | 11 |
| RSPH4A | NG_012934.1 | NM_001010892.2 | NP_001010892.1 | 7 |
| RSPH9 | NG_023436.1 | NM_152732.4 | NP_689945.2 | 7 |
| SPAG1 | NG_033834.1 | NM_172218.2 | NP_757367.1 | 21 |
| TEKT1 | NC_000017.11 | NM_053285.1 | NP_444515.1 | 8 |
| TTC25 | NG_053115.1 | NM_031421.3 | NP_113609.1 | 13 |
| ZMYND10 | NG_042828.1 | NM_015896.2 | NP_056980.2 | 12 |
ID data were obtained from the National Center for Biotechnology Information (NCBI; https://www.ncbi.nlm.nih.gov/) database.
ID: identification.
Regions of interest were captured following the commercial protocol (SeqCap EZ [Roche NimbleGen, Pleasanton, CA, USA]), with 21 min enzymatic fragmentation. The library was sequenced using a next-generation MiSeq benchtop sequencer (Illumina, San Diego, CA, USA). The data analysis process included trimming the sequences with Trimmomatic (Institute for Biology, Aachen, Germany),15 aligning the sequences with the reference human genome GRCh (hg38) using BWA-MEM,16 detecting variants with the Genome Analysis Toolkit (GATK) Haplotype Caller (Broad Institute, Cambridge, MA, USA),17 and annotating variants with ANNOVAR.18 Variants with a coverage less than 20 were not considered in the analysis. The list of identified variants was compared with information from specific databases to identify variants already found to be associated with a known phenotype (HGMD, ClinVar) and population frequency databases (GnomAD, ExAC, 1000 genomes) to rule out variants that are present in the general population at a rate higher than 1%. In parallel, data were also analyzed using VariantStudio v2.2.1 (Illumina®, San Diego, CA, USA). The pathogenicity of the variants was evaluated using Alamut v2.11 software (Interactive Biosoftware, Rouen, France) which includes Mutation Taster, Polyphen, Aling GVGD and SIFT, and Varsome (Saphetor, Lausanne, Switzerland) which includes DANN, Gerp and MutationTaster. The effect of mutations identified in splicing regions was evaluated by SpliceSiteFinder, MaxEntScan, NNSPLICE, GeneSplicant, and Human Splicing Finder, also included in Alamut v2.11. Massive sequencing data were reanalyzed on ExomeDepth,19 a bioinformatic platform used to detect copy number variations (CNV). Nomenclature and classification of variants are based on guidelines from the Human Genome Variation Society (HGVS) (https://www.hgvs.org/)20 and the American College of Medical Genetics and Genomics (ACMGG) (https://www.acmg.net/).21
Probable pathogenic variants were confirmed in patients using Sanger sequencing and, where possible, familial cosegregation was analyzed.
Statistical analysisThe percentage, median and range, and mean and standard deviation (SD) were used for the description of the variables. To calculate the sensitivity and specificity of the gene panel, cases of confirmed or highly probable PCD were considered as cases diagnosed with PCD. The Chi-squared test was used for comparison between adult and child patients, with a p value < 0.05 being statistically significant. Analyses were conducted using MedCalc Statistical Software version 19.1.3 (MedCalc Software bvba, Ostende, Belgium).
ResultsBetween January 2017 and November 2019, 79 patients from 74 different families (74 index cases) and 39 family members were studied. Of the 79 patients, 26 were classified as very unlikely PCD and in all of them the genetic study was negative.
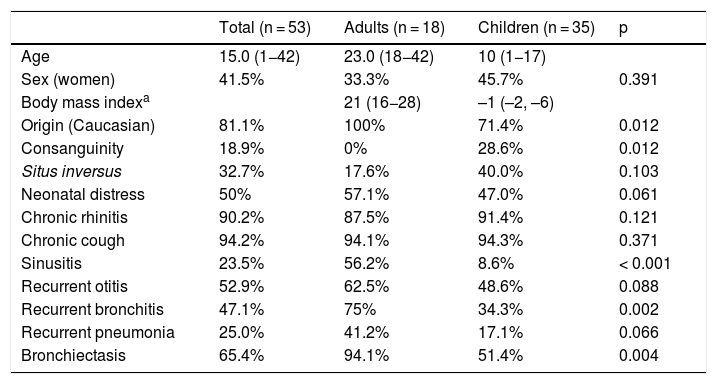
Of the 53 patients with confirmed or highly probable diagnosis of PCD, 35 were children and 18 were adults. Forty-three patients were Caucasian, 4 (7.5%) Moroccan, 4 (7.5%) Pakistani, 1 was from the Middle East, and 1 from Latin America. Ten patients had a family history of consanguinity (Table 2 and Appendix B Table 1S, supplemental material). The most common clinical manifestations were chronic cough and chronic rhinitis. Half of the series had a history of neonatal distress and 32.7% had situs inversus. The frequency of bronchiectasis was higher in adult patients (94.1%) than in pediatric patients (51.4%) (Table 2 and Appendix B Table 1S, supplementary material). The PICADAR score was equal to or greater than 5 in 31 patients (65.9%). The value of nNO could be determined in 35 cases, with an average value of 25.9 (SD 29.1) nl/min. In 25 patients, it was less than 33 nl/min and in only 2 was it more than 77 nl/min (Appendix B Table 1S, supplementary material). HSVM and EM findings are listed in Appendix B Table 1S, supplementary material. In 15 cases, the alteration observed on EM was considered diagnostic. HSVM was highly indicative of PCD in 52 patients (not available in patient 3), with the following alterations being found: static pattern (n = 18), static pattern with residual motion (n = 12), stiff, disorganized pattern (n = 8), hyperkinetic pattern (n = 5), rotating pattern (n = 6), dyskinesia (n = 2), and reduced distal movement (n = 1).
Clinical characteristics of patients with primary ciliary dyskinesia included in the study.
| Total (n = 53) | Adults (n = 18) | Children (n = 35) | p | |
|---|---|---|---|---|
| Age | 15.0 (1−42) | 23.0 (18−42) | 10 (1−17) | |
| Sex (women) | 41.5% | 33.3% | 45.7% | 0.391 |
| Body mass indexa | 21 (16−28) | –1 (–2, –6) | ||
| Origin (Caucasian) | 81.1% | 100% | 71.4% | 0.012 |
| Consanguinity | 18.9% | 0% | 28.6% | 0.012 |
| Situs inversus | 32.7% | 17.6% | 40.0% | 0.103 |
| Neonatal distress | 50% | 57.1% | 47.0% | 0.061 |
| Chronic rhinitis | 90.2% | 87.5% | 91.4% | 0.121 |
| Chronic cough | 94.2% | 94.1% | 94.3% | 0.371 |
| Sinusitis | 23.5% | 56.2% | 8.6% | < 0.001 |
| Recurrent otitis | 52.9% | 62.5% | 48.6% | 0.088 |
| Recurrent bronchitis | 47.1% | 75% | 34.3% | 0.002 |
| Recurrent pneumonia | 25.0% | 41.2% | 17.1% | 0.066 |
| Bronchiectasis | 65.4% | 94.1% | 51.4% | 0.004 |
The data are expressed as median and range (in brackets) for quantitative variables (age, body mass index) and as a percentage for qualitative variables.
DNA samples were sequenced using our gene panel, which covered 98.75% of the exons and flanking intronic areas of the 44 genes included (Table 1). The average coverage of the results was 600x with 80.7% reads on target.
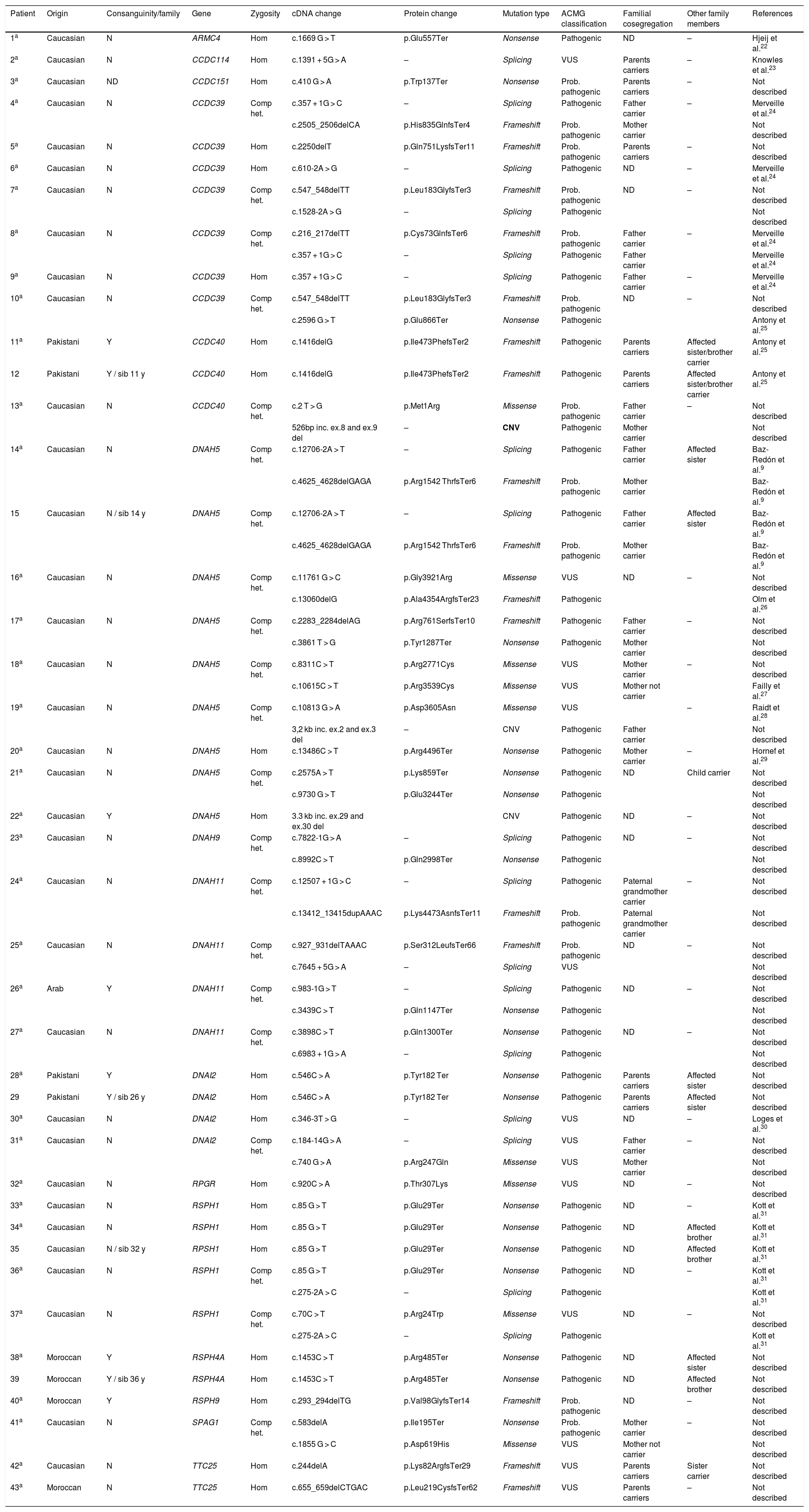
Candidate variants were found in 81.1% (43/53) of patients with PCD in some of the panel genes, with 22 (51.2%) homozygous and 21 (48.8%) compound heterozygous. In 18.9% (10/53) of the patients, no variant was found that could explain the phenotype (Table 3). The sensitivity of the technique was 81.1% (95% CI, 68.0%-90.6%) and specificity was 100% (95% CI, 86.8%-100%). The area under the ROC curve was 0.91 (95% CI, 0.82−0.96). The positive predictive value of the gene panel in our study population, where the prevalence of PCD cases was 67.1%, was 100% and the negative predictive value was 72.2% (95% CI, 59.8%-82.0%). A total of 52 different variants were found (1 in ARMC4, 1 in CCDC114, 1 in CCDC151, 8 in CCDC39, 3 in CCDC40, 14 in DNAH5, 2 in DNAH9, 8 in DNAH11, 4 in DNAI2, 1 in RPGR, 3 in RSPH1, 1 in RSPH4A, 1 in RSPH9, 2 in SPAG1, and 2 in TTC25), 16 of which had previously been associated with PCD9,22–31 and 36 that had not been previously described in the literature (Table 3). Of the 52 variants found, 14 (26.9%) were nonsense variants, 13 (25%) were frameshift, 13 (25%) splicing, 9 (17.3%) missense, and 3 (5.8%) CNVs. Overall, 51.9% (27/52) were classified as pathogenic (including the 3 CNVs), 21.2% (11/52) as probably pathogenic, and 26.9% (14/52) as variants of uncertain significance (VUS), according to the ACMG classification (Table 3).
Results of the genetic study of patients with described variants that correlate with their phenotype.
| Patient | Origin | Consanguinity/family | Gene | Zygosity | cDNA change | Protein change | Mutation type | ACMG classification | Familial cosegregation | Other family members | References |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1a | Caucasian | N | ARMC4 | Hom | c.1669 G > T | p.Glu557Ter | Nonsense | Pathogenic | ND | – | Hjeij et al.22 |
| 2a | Caucasian | N | CCDC114 | Hom | c.1391 + 5G > A | – | Splicing | VUS | Parents carriers | – | Knowles et al.23 |
| 3a | Caucasian | ND | CCDC151 | Hom | c.410 G > A | p.Trp137Ter | Nonsense | Prob. pathogenic | Parents carriers | – | Not described |
| 4a | Caucasian | N | CCDC39 | Comp het. | c.357 + 1G > C | – | Splicing | Pathogenic | Father carrier | – | Merveille et al.24 |
| c.2505_2506delCA | p.His835GlnfsTer4 | Frameshift | Prob. pathogenic | Mother carrier | Not described | ||||||
| 5a | Caucasian | N | CCDC39 | Hom | c.2250delT | p.Gln751LysfsTer11 | Frameshift | Prob. pathogenic | Parents carriers | – | Not described |
| 6a | Caucasian | N | CCDC39 | Hom | c.610-2A > G | – | Splicing | Pathogenic | ND | – | Merveille et al.24 |
| 7a | Caucasian | N | CCDC39 | Comp het. | c.547_548delTT | p.Leu183GlyfsTer3 | Frameshift | Prob. pathogenic | ND | – | Not described |
| c.1528-2A > G | – | Splicing | Pathogenic | Not described | |||||||
| 8a | Caucasian | N | CCDC39 | Comp het. | c.216_217delTT | p.Cys73GlnfsTer6 | Frameshift | Prob. pathogenic | Father carrier | – | Merveille et al.24 |
| c.357 + 1G > C | – | Splicing | Pathogenic | Father carrier | Merveille et al.24 | ||||||
| 9a | Caucasian | N | CCDC39 | Hom | c.357 + 1G > C | – | Splicing | Pathogenic | Father carrier | – | Merveille et al.24 |
| 10a | Caucasian | N | CCDC39 | Comp het. | c.547_548delTT | p.Leu183GlyfsTer3 | Frameshift | Prob. pathogenic | ND | – | Not described |
| c.2596 G > T | p.Glu866Ter | Nonsense | Pathogenic | Antony et al.25 | |||||||
| 11a | Pakistani | Y | CCDC40 | Hom | c.1416delG | p.Ile473PhefsTer2 | Frameshift | Pathogenic | Parents carriers | Affected sister/brother carrier | Antony et al.25 |
| 12 | Pakistani | Y / sib 11 y | CCDC40 | Hom | c.1416delG | p.Ile473PhefsTer2 | Frameshift | Pathogenic | Parents carriers | Affected sister/brother carrier | Antony et al.25 |
| 13a | Caucasian | N | CCDC40 | Comp het. | c.2 T > G | p.Met1Arg | Missense | Prob. pathogenic | Father carrier | – | Not described |
| 526bp inc. ex.8 and ex.9 del | – | CNV | Pathogenic | Mother carrier | Not described | ||||||
| 14a | Caucasian | N | DNAH5 | Comp het. | c.12706-2A > T | – | Splicing | Pathogenic | Father carrier | Affected sister | Baz-Redón et al.9 |
| c.4625_4628delGAGA | p.Arg1542 ThrfsTer6 | Frameshift | Prob. pathogenic | Mother carrier | Baz-Redón et al.9 | ||||||
| 15 | Caucasian | N / sib 14 y | DNAH5 | Comp het. | c.12706-2A > T | – | Splicing | Pathogenic | Father carrier | Affected sister | Baz-Redón et al.9 |
| c.4625_4628delGAGA | p.Arg1542 ThrfsTer6 | Frameshift | Prob. pathogenic | Mother carrier | Baz-Redón et al.9 | ||||||
| 16a | Caucasian | N | DNAH5 | Comp het. | c.11761 G > C | p.Gly3921Arg | Missense | VUS | ND | – | Not described |
| c.13060delG | p.Ala4354ArgfsTer23 | Frameshift | Pathogenic | Olm et al.26 | |||||||
| 17a | Caucasian | N | DNAH5 | Comp het. | c.2283_2284delAG | p.Arg761SerfsTer10 | Frameshift | Pathogenic | Father carrier | – | Not described |
| c.3861 T > G | p.Tyr1287Ter | Nonsense | Pathogenic | Mother carrier | Not described | ||||||
| 18a | Caucasian | N | DNAH5 | Comp het. | c.8311C > T | p.Arg2771Cys | Missense | VUS | Mother carrier | – | Not described |
| c.10615C > T | p.Arg3539Cys | Missense | VUS | Mother not carrier | Failly et al.27 | ||||||
| 19a | Caucasian | N | DNAH5 | Comp het. | c.10813 G > A | p.Asp3605Asn | Missense | VUS | – | Raidt et al.28 | |
| 3,2 kb inc. ex.2 and ex.3 del | – | CNV | Pathogenic | Father carrier | Not described | ||||||
| 20a | Caucasian | N | DNAH5 | Hom | c.13486C > T | p.Arg4496Ter | Nonsense | Pathogenic | Mother carrier | – | Hornef et al.29 |
| 21a | Caucasian | N | DNAH5 | Comp het. | c.2575A > T | p.Lys859Ter | Nonsense | Pathogenic | ND | Child carrier | Not described |
| c.9730 G > T | p.Glu3244Ter | Nonsense | Pathogenic | Not described | |||||||
| 22a | Caucasian | Y | DNAH5 | Hom | 3.3 kb inc. ex.29 and ex.30 del | CNV | Pathogenic | ND | – | Not described | |
| 23a | Caucasian | N | DNAH9 | Comp het. | c.7822-1G > A | – | Splicing | Pathogenic | ND | – | Not described |
| c.8992C > T | p.Gln2998Ter | Nonsense | Pathogenic | Not described | |||||||
| 24a | Caucasian | N | DNAH11 | Comp het. | c.12507 + 1G > C | – | Splicing | Pathogenic | Paternal grandmother carrier | – | Not described |
| c.13412_13415dupAAAC | p.Lys4473AsnfsTer11 | Frameshift | Prob. pathogenic | Paternal grandmother carrier | Not described | ||||||
| 25a | Caucasian | N | DNAH11 | Comp het. | c.927_931delTAAAC | p.Ser312LeufsTer66 | Frameshift | Prob. pathogenic | ND | – | Not described |
| c.7645 + 5G > A | – | Splicing | VUS | Not described | |||||||
| 26a | Arab | Y | DNAH11 | Comp het. | c.983-1G > T | – | Splicing | Pathogenic | ND | – | Not described |
| c.3439C > T | p.Gln1147Ter | Nonsense | Pathogenic | Not described | |||||||
| 27a | Caucasian | N | DNAH11 | Comp het. | c.3898C > T | p.Gln1300Ter | Nonsense | Pathogenic | ND | – | Not described |
| c.6983 + 1G > A | – | Splicing | Pathogenic | Not described | |||||||
| 28a | Pakistani | Y | DNAI2 | Hom | c.546C > A | p.Tyr182 Ter | Nonsense | Pathogenic | Parents carriers | Affected sister | Not described |
| 29 | Pakistani | Y / sib 26 y | DNAI2 | Hom | c.546C > A | p.Tyr182 Ter | Nonsense | Pathogenic | Parents carriers | Affected sister | Not described |
| 30a | Caucasian | N | DNAI2 | Hom | c.346-3T > G | – | Splicing | VUS | ND | – | Loges et al.30 |
| 31a | Caucasian | N | DNAI2 | Comp het. | c.184-14G > A | – | Splicing | VUS | Father carrier | – | Not described |
| c.740 G > A | p.Arg247Gln | Missense | VUS | Mother carrier | Not described | ||||||
| 32a | Caucasian | N | RPGR | Hom | c.920C > A | p.Thr307Lys | Missense | VUS | ND | – | Not described |
| 33a | Caucasian | N | RSPH1 | Hom | c.85 G > T | p.Glu29Ter | Nonsense | Pathogenic | ND | – | Kott et al.31 |
| 34a | Caucasian | N | RSPH1 | Hom | c.85 G > T | p.Glu29Ter | Nonsense | Pathogenic | ND | Affected brother | Kott et al.31 |
| 35 | Caucasian | N / sib 32 y | RPSH1 | Hom | c.85 G > T | p.Glu29Ter | Nonsense | Pathogenic | ND | Affected brother | Kott et al.31 |
| 36a | Caucasian | N | RSPH1 | Comp het. | c.85 G > T | p.Glu29Ter | Nonsense | Pathogenic | ND | – | Kott et al.31 |
| c.275-2A > C | – | Splicing | Pathogenic | Kott et al.31 | |||||||
| 37a | Caucasian | N | RSPH1 | Comp het. | c.70C > T | p.Arg24Trp | Missense | VUS | ND | – | Not described |
| c.275-2A > C | – | Splicing | Pathogenic | Kott et al.31 | |||||||
| 38a | Moroccan | Y | RSPH4A | Hom | c.1453C > T | p.Arg485Ter | Nonsense | Pathogenic | ND | Affected sister | Not described |
| 39 | Moroccan | Y / sib 36 y | RSPH4A | Hom | c.1453C > T | p.Arg485Ter | Nonsense | Pathogenic | ND | Affected brother | Not described |
| 40a | Moroccan | Y | RSPH9 | Hom | c.293_294delTG | p.Val98GlyfsTer14 | Frameshift | Prob. pathogenic | ND | – | Not described |
| 41a | Caucasian | N | SPAG1 | Comp het. | c.583delA | p.Ile195Ter | Nonsense | Prob. pathogenic | Mother carrier | – | Not described |
| c.1855 G > C | p.Asp619His | Missense | VUS | Mother not carrier | Not described | ||||||
| 42a | Caucasian | N | TTC25 | Hom | c.244delA | p.Lys82ArgfsTer29 | Frameshift | VUS | Parents carriers | Sister carrier | Not described |
| 43a | Moroccan | N | TTC25 | Hom | c.655_659delCTGAC | p.Leu219CysfsTer62 | Frameshift | VUS | Parents carriers | – | Not described |
–: data missing; ACMG: American College of Medical Genetics; bp: base pairs; Comp het.: compound heterozygote; Ex.: exon; Het: heterozygote; Hom: homozygote; kb: kilobases; ND: no data; Prob. pathogenic: probably pathogenic; sib: sibling; VUS: variant of uncertain significance; y: years of age.
Eighteen patients presented variants in genes related to structural proteins of the outer dynein arms (DNAH5 [n = 9], DNAH11 [n = 4], DNAI2 [n = 4], DNAH9 [n = 1]) and 5 related to the outer dynein arm docking complex (TTC25 [n = 2], ARMC4 [n = 1], CCDC114 [n = 1], CCDC151 [n = 1]); 8 showed variants in genes encoding radial spoke proteins (RSPH1 [n = 5], RSPH4A [n = 2], RSPH9 [n = 1]); in 10, variants were detected in genes encoding axoneme regulatory complex proteins (CCDC39 [n = 7], CCDC40 (n = 3]); 1 patient presented variants in SPAG1, which encodes a protein probably related to the transport or cytoplasmic assembly of dynein complexes, and 1 with variants in RPGR, a gene associated with retinitis pigmentosa (Fig. 1, Table 3). The variants in the 3 most common genes (DNAH5, CCDC39 and RSPH1) occurred only in patients of Caucasian origin. In patients of non-Caucasian origin, the most common causative genes were CCDC40, DNAI2 and RSPH4A, with 2 cases each.

Cross-sectional diagram of a respiratory cilium showing its structural components and genes in which variants have been found. The number of patients with variants in each gene is shown in parentheses. N-DRC: nexin-dynein regulator complex; Outer dynein arm-DC: outer dynein arm docking complex.
Thirty-seven family members from 22 different families have been tested using the gene panel. All parents analyzed (18 different families) were carriers of some of the variants found in their children. DNA from the maternal grandparents and the maternal grandmother of patient 24 was analyzed and the variant c.12507 + 1G > C was determined to be of paternal origin, while the variant c.13412_13415dupAAAC was of maternal origin (Table 3).
DiscussionIn a cohort of 53 patients with confirmed or highly probable PCD, positive genetic results were obtained in 81.1% of cases using a massive sequencing panel of 44 genes, allowing us to identify the gene causing the ciliary structure defect. In another 26 patients referred for suspicious respiratory symptoms, but with a diagnosis of very unlikely PCD after initial tests, the genetic study was negative. This is the first study, to our knowledge, to describe the genes that cause ciliary dyskinesia in a large cohort of patients in Spain.
Our results have confirmed that, in our population, this gene panel has a high diagnostic yield (sensitivity of 81.1%) and that it was able to rule out all patients with a low suspicion of PCD (specificity of 100%). The yield of gene panels applied to other populations has increased as new genes are discovered and incorporated into panels, and now ranges between 43% and 70%32–34 and more recently 82%.35
The diagnosis of PCD using the techniques available to date is complex and generates many diagnostic uncertainties and doubts among doctors and patients concerning the prognosis and course of the disease. The determination of nNO with a cut-off point 77 nl/min has high sensitivity (93.6%), but a specificity of 78.9%.36 Electron microscopy is specific (100%), but fails to identify 21% of cases. Furthermore, it must be interpreted by highly skilled operators, and suitable samples are not always obtained.5,37 HSVM has excellent sensitivity and specificity; however, it also needs expert technicians and often has to be repeated several times.8 Although genetic studies may fail to identify 20% of cases, they yield a definitive diagnosis and provide clearer guidance for treatment and genetic counselling, and a better groundwork for research into specific treatments, such as gene therapy or protein therapies.13
Massive sequencing with the gene panel can be used to study specific variants and small deletions or insertions (indels) and copy number variants (CNVs) of genes described to date as causing PCD. With this technique, a proportion of patients with confirmed or highly probable PCD, 18.9% in our series, is left without a genetic diagnosis. In these patients, analysis of the entire exome may help identify new genes that cause PCD.
The majority of the variants described in our patients (82.7%) caused loss of protein function (nonsense, frameshift, CNV and splicing), similar to results described in other studies.35 In 9 (17.3%) patients, we found missense variants involving the change of a single amino acid that were catalogued as VUS according to the ACMG 21 classification, with the exception of the c.2 T > G/p.Met1Arg variant (patient 13) that affected the first amino acid and was classified as probably pathogenic (Table 3). These missense variants were taken into account as a possible cause of protein alteration according to in silico predictions. Ideally, these missense defects should be tested in vitro in patient nasal respiratory epithelium cell cultures or animal models.
Given the large number of variants that can be identified with massive sequencing, many of which are benign, results can only be interpreted correctly if variants identified correlate with EM and HSVM findings. In our series, a good correlation between ultrastructure and genetic findings was obtained in only 6 cases, given the difficulties of EM interpretation and the possibilities of changes due to respiratory infections or processing artifacts,9,37 while the HSVM study showed a good correlation with genetic findings in all cases.
Patients 13 and 19, who initially had a single heterozygous variant in genes CCDC40 and DNAH5, respectively, were resolved with a bioinformatics analysis of CNV, as was the case in patient 22. In patients 13 and 19, deletions were described in the other allele that also agreed with the familial segregation study, with the mother of patient 13 and the father of patient 19 being identified as the carriers of these deletions (Table 3). In patient 22, a homozygous deletion was detected in the DNAH5 gene. Bioinformatics analysis of CNV is a useful tool for resolving some cases, especially those with monoallelic variants in a candidate gene that fits the phenotype.
It should be noted that all cases of consanguinity in our cohort had a positive molecular result and were homozygous for the variants found, all of which were classified as pathogenic or probably pathogenic (Table 3).
The distribution of genes that cause PCD differs depending on ethnicity.35 In our series, DNAH5 and CCDC39 were the most prevalent genes and both were found only in patients of Caucasian origin (Table 3). DNAH5 has been described as the most frequent gene in Caucasian studies, explaining 15%-37% of cases,27,32,33,35 but it is rare in other populations, such as Arabs.34,35 The CCDC39 gene has previously been described in patients of European origin25 and is one of the predominant genes in the population of Arab origin.34,35
The limitations of our study are mainly related to the number of patients studied which, although significant for a rare disease, must be expanded to better understand the frequency of the different variants in our population, both Caucasian and non-Caucasian. Massive sequencing cannot detect all deletions/duplications in genes, but this issue has been solved by bioinformatics processing with CNV analysis, although this is only an approximation and it is advisable to confirm findings with other methods. Other limitations are those inherent to gene panel studies, since neither the entire exome and nor the genome are analyzed. However, this facilitates the interpretation of results, since the analysis of the whole exome or genome may contain a very high number of variants that lack pathogenic significance in the healthy population. In gene panels, moreover, coverage is optimized with respect to exome sequencing. Although our custom gene panel has made it possible to determine the specific defect of patients diagnosed using molecular techniques, new PCD genes are described every year,38–40 so this panel must be expanded with recently discovered data.
In conclusion, the results of this study show the utility of designing and implementing genetic analysis using custom gene panels, which is a useful tool for improving the diagnosis of PCD.
FundingThis study was funded by a Strategic Action in Health grant from the Instituto de Salud Carlos III (ISCIII) (PI16/01233), co-funded by the European Regional Development Fund, Operational Program Smart Growth 2014–2020, a grant from the Spanish Society of Pediatric Pulmonology (2016), and a scholarship from the Catalan Pulmonology Foundation (FUCAP) (2016). NCT received support for a Short Term Scientific Mission from COST ActionBM1407.
Conflict of interestsThe authors state that they have no conflict of interests.
The authors participate in COST Action BM1407 Translational research in primary ciliary dyskinesia: bench, bedside, and population perspectives (BEAT PCD). AMG and SRA participate in ERN-LUNG. This study was part of the Pediatrics, Obstetrics and Gynecology doctoral program at the Universitat Autònoma de Barcelona. We thank Dr. Josep Quer and Damir García-Cehic (Vall d’Hebron Institut de Recerca [VHIR], Barcelona) for their valuable collaboration and for their excellent assistance in massive sequencing techniques.
The following are Supplementary data to this article:
Please cite this article as: Baz-Redón N, Rovira-Amigo S, Paramonov I, Castillo-Corullón S, Cols-Roig M, Antolín M, et al. Implementación de un panel de genes para el diagnóstico genético de la discinesia ciliar primaria. Arch Bronconeumol. 2021;57:186–194.




www.publicationethics.org.




Archivos de Bronconeumología follows the Recommendations for the Conduct, Reporting, Editing and Publication of Scholarly Work in Medical Journals