

Cystic fibrosis (CF) is the most frequent life-shortening inherited disease in Caucasians, caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene.1 Loss or altered CFTR-function results in impaired transport of chloride and sodium, bicarbonate and water to the lumen of exocrine glands in different organ systems. Consequently, viscous secretions impair mucociliary clearance, and facilitate pathogen colonization, leading to pulmonary destruction and premature death in almost 90% of CF-patients. Furthermore, the majority of CF-patients reveal abdominal involvement with pancreatic insufficiency (PI), intestinal and hepatic manifestations. The recent discovery of small molecules modulating CFTR-dysfunction brought a major breakthrough in therapy of CF in the CFTR-gating mutation G551D, which worldwide concerns only 2%–4% of patients.2–4 The small molecule VX-770 (ivacaftor/IVA) increases Cl−-secretion in human CF bronchial epithelia carrying G551D to almost 50% of the potential without CF.5 Consequently, VX-770 prompted the highest clinical effects hitherto achieved with a CF-drug, improving pulmonary function (FEV1) for 10%–12%, stabilizing weight and reducing sweat chloride from about 100mmol/l to about 50mmol/l.6–8 The drug was approved for adult and pediatric CF patients carrying a G551D mutation, and only recently down to the age of 2 years.2,3,9
To the best of our knowledge, this is the first report on three siblings with CF carrying the rare G551D-mutation and receiving the novel drug. The siblings reveal different symptoms and severity of disease manifestation, with intermittent Pseudomonas aeruginosa (PSA) colonization of the second child's airways and diverging grades of pancreatic insufficiency (PI). However, until now they all present normal pulmonary function before and during the newly introduced CFTR-modulating therapy. Nevertheless, we show how the potent CFTR-modulator for gating mutations effectively improves involvement of the different organ systems.
Case A (female, *03.2005). The families’ second child (Sib.2), was first diagnosed with CF. At the age of 5 years recurrent pneumonia, voluminous foul-smelling stools and weight below the 3rd percentile prompted a sweat test, which resulted in chloride values of 121mmol/l. One week later CF diagnosis was confirmed with detection of the CFTR-mutations G551D and F508del. The patient was intermittently colonized with PSA before initiation of IVA, and luckily the critical pathogen could be eradicated. Already at diagnosis she apparently resulted to be PI revealing pancreas elastase in stool of 54μg/g (borderline ≥200/normal ≥500) (see Fig. 1). Despite supplementation of pancreatic enzymes since the days of diagnosis, her weight remained about the 10th percentile. Together with her elder brother she receives ivacaftor since 01.2013, which impressively reduced sweat chloride down to normal (26.4mmol/l) and pancreatic elastase increased to 189μg/g of stool. However, she did not reveal a significant increase of pulmonary function, which before and after initiation of the CFTR-modulator varies between 89 and 118 percent predicted (FEV1), and her BMI-percentiles, which initially increased from the 27th to the 36th percentile now declined to the 14th percentile, which we attribute to low caloric and enzyme intake despite pancreatic insufficiency.
 with ivacaftor (IVA) (<200μg/g of stool: reduced elastase; 200 to <500μg/g: borderline values; ≥500μg/g: normal).")
Case B (male, *09.2003). At the day of the younger sisters’ CF-diagnosis her parents were questioned for ‘salty taste when kissing her’. They confirmed but stated, that also the older brother (Sib.1) revealed this symptom. Thus, the 7-year old boy with a normal growth pattern was confirmed to carry the same CFTR-mutations G551D and F508del. Retrospectively, the parents reported recurrent constipation and chronic rhinosinusitis which had required adenoidectomy. For allergic sensitization to pollen and atopic dermatitis these had been classified as atopic symptoms. At presentation, his sweat test resulted highly positive with 121mmol/l and Haemophilus parainfluenza and Staphylococcus aureus were isolated from his oropharyngeal swabs (see Table 1). Abdominal ultrasound (US) showed increased echogenicity of the pancreas and liver as well as a single large gallstone. Interestingly, the patient nevertheless was pancreatic sufficient (PS) with pancreas elastase of 343μg/g, which six months later declined to 278μg/g (see Fig. 1) despite normal serum amylase and lipase levels during the follow-ups. Initiation of ivacaftor in January 2013 led to normalization of pancreatic elastase (>500μm/g) and pulmonary function always resulted to be normal without a further significant improvement during treatment with the CFTR modulator.
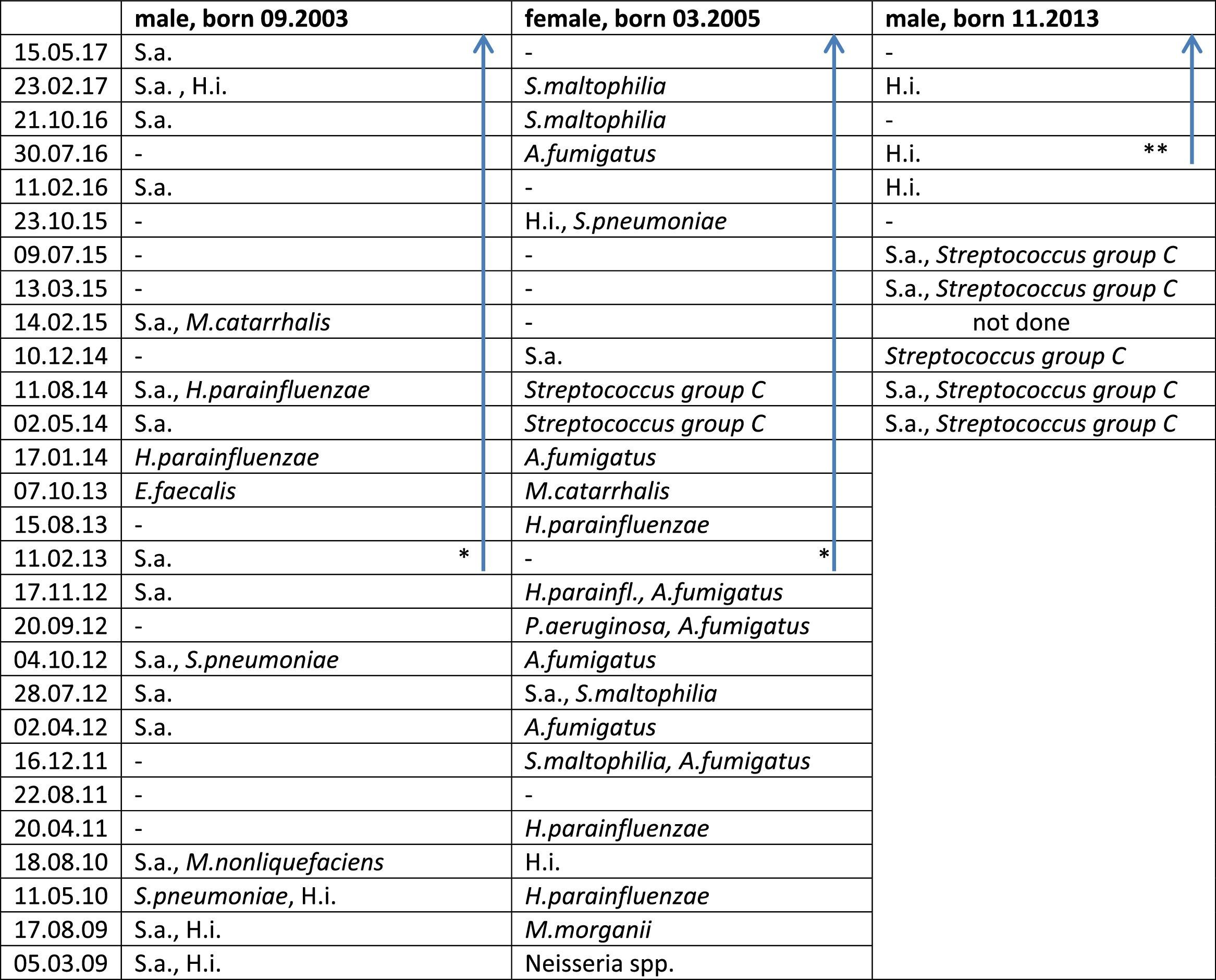
Lower airway Pathogen Colonization in the Three Siblings Before And During Therapy With Ivacaftor.
* Start ivacaftor 01.01.2013.
** Start ivacaftor 20.04.2016; blue arrow: during treatment with ivacaftor.
Abbreviations – P. aeruginosa: Pseudomonas aeruginosa; S.a.: Staphylococcus aureus; H.i.: Haemophilus influenzae; H. parainfluenzae: Haemophilus parainfluenzae; S. pneumoniae: Streptococcus pneumoniae; S. maltophilia: Stenotrophomonas maltophiliae; A. fumigatus: Aspergillus fumigatus; M. morganii: Morganella morganii; M. nonliquefaciens: Moraxella nonliquefaciens; E. faecalis: Enterococcus faecalis; A. fumigatus: Aspergillus fumigatus; –: no pathogens detected.
Case C (male, *11.2013). The third sibling (Sib.3) was born after an uncomplicated gestation. For the elder siblings’ good course despite suffering from CF the parents had decided against prenatal genetic diagnostics. However, abdominal sonography during the third-trimester showed elevated echogenicity of the fetus’ small bowel. Still, he passed a normal meconium within 24hours after birth. Although the child initially thrived normally without pancreatic enzyme substitution, genetic analysis revealed CF with compound heterozygoty for G551D and F508del. Interestingly, pancreas elastase as a newborn resulted normal at 470μg/g but it gradually declined to levels of pancreatic insufficiency (see Fig. 1), while lipase in serum increased to 3.3μmol/ls (normal<1.33), revealing acute pancreatic damage. Nevertheless the childs’ weight remained about the 50th percentile but he required substitution of pancreatic enzymes before the age of 2 years, when ivacaftor was introduced. Interestingly, pancreatic elastase in stool then increased almost to borderline values (181μg/g) (see Fig. 1).
This is the first report on three siblings suffering from CF, carrying the rare G551D-mutation together with F508del, the most common mutation in Europe and receiving therapy with the new CFTR-modulator ivacaftor. Altogether, heterogeneity of the siblings’ pancreatic phenotype is noticeable, revealing a certain degree of PI in siblings 2 and 3 whereas sibling 1 still remains PS. Remarkably, all siblings showed a tendency to recover some pancreatic function during treatment with ivacaftor. This accords well to the recent report from Davies et al. on CF-patients aged 2–5 years carrying a G551D mutation: some of them also recovered pancreatic function during a newly introduced ivacaftor-treatment.9
However, we suggest that at least sibling 2 would require a more intensive nutritional support and enzyme substitution, more accordingly to classical CF, in order to stabilize weight and thriving.
Concluding, our report on three siblings with the rare CFTR-mutation G551D reveals a different severity of disease manifestation but altogether a normal pulmonary function. In the siblings introduction of the CFTR-modulator ivacaftor did not relevantly improve FEV1 or the BMI-percentiles. However, we demonstrate with sweat tests and pancreatic elastase that the modulator effectively improves the multi-organ disease. Therefore we suppose that on the long run the CFTR-modulator should reduce disease progression with gastrointestinal involvement and pulmonary destruction as major reason for premature death with the inherited disease.
Conflicts of InterestsJGM reports a study grant from Vertex (IIT) as well as personal fees from Vertex for Advisory Boards and Lectures, outside the submitted work.
The authors thank the CF family for their support in this project.







