



El déficit de alfa-1 antitripsina (DAAT) es un trastorno autosómico codominante que predispone al desarrollo temprano de enfermedad pulmonar obstructiva crónica (EPOC) y/o patología hepática. Se considera que una concentración plasmática de alfa-1 antitripsina (ATT) por debajo de 50mg/dl representa una deficiencia relevante y es causada por la herencia de 2 alelos de deficiencia grave del gen SERPINA11. El 95% de los casos clínicos relacionados con dicho déficit están asociados al genotipo PI*ZZ, siendo el 5% restante correspondiente a los genotipos PI*SZ, PI*MZ o a combinaciones de PI*S o PI*Z con otros alelos deficitarios o nulos extremadamente raros. Estos alelos raros representan el 4,6% de las variantes deletéreas anotadas en el Registro Español de Pacientes con Déficit de AAT, siendo las variantes nulas muy poco frecuentes2. Si bien en las 2 últimas décadas se han descubierto alrededor de 25 variantes nulas, existe escasa información acerca de su impacto clínico3-9. Presentamos el caso de 2 pacientes remitidos a la consulta de neumología con diagnóstico de DAAT en relación con el alelo PI*Q0amersfoort y PI*Q0cardiff.
El primer caso es un mujer de 47 años de edad, natural de la antigua Yugoslavia, con antecedentes personales de tabaquismo y un índice paquetes-año de 15, que es remitida a la consulta de neumología por disnea a moderados esfuerzos (mMRC 2) de un año de evolución. Las pruebas funcionales respiratorias fueron las siguientes: FEV1/FVC 0,5; FEV1 1,80 l (55%); FVC 3,40 l (77%); DLCO 53%; KCO 52%. La TAC de alta resolución describía la presencia de enfisema centrolobulillar de predominio en ambos lóbulos inferiores. El hemograma, niveles de IgA, IgM, IgG, IgE y de transaminasas se encontraban dentro de la normalidad. La medida de AAT plasmática por nefelometría fue de 18mg/dl, por lo que se propuso hacer estudio genético dirigido a alelos deficitarios S y Z mediante PCR a tiempo real (sondas FRET, LightCycler 2.0, TIB MOLBIOL) que detectó la presencia de un alelo PI*Z en heterocigosis, siendo descartado la presencia de alelos PI*S. Ante la discordancia entre el genotipo obtenido y los niveles plasmáticos de AAT, se decide realizar el estudio molecular de todas las regiones exónicas codificantes y de las zonas intrónicas flanqueantes del gen SERPINA1 mediante secuenciación Sanger (BigDye™ Terminator v3.1 Cycle Sequencing, Thermo Fisher Scientific). En dicho estudio, además de la variante PI*Z anteriormente descrita, se detectó la presencia en heterocigosis del alelo PI*Q0amersfoort (fig. 1).
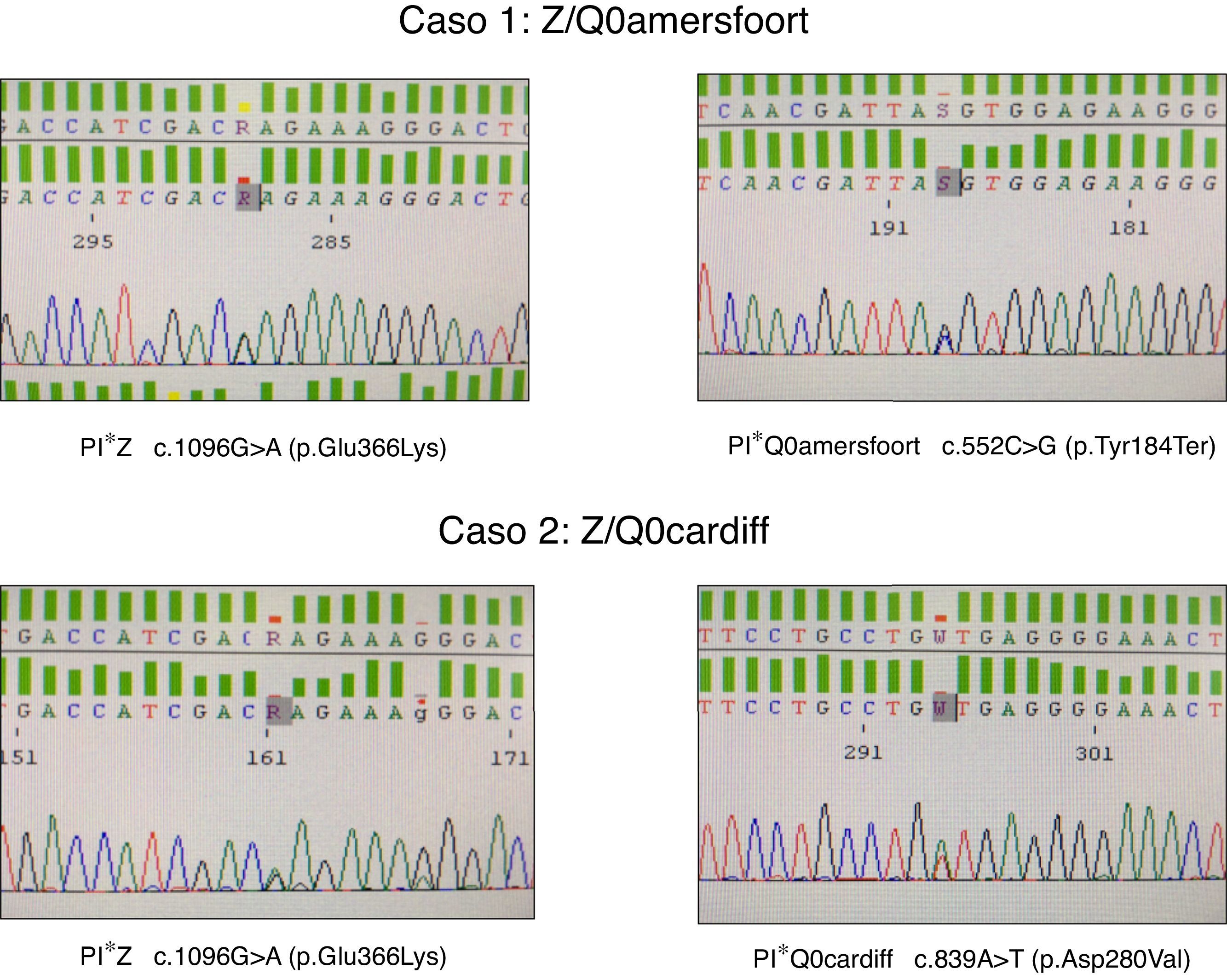
El segundo caso es un varón de 42 años de edad y sin antecedentes personales de interés, que es remitido a nuestra consulta por historia familiar de DAAT. Presenta un déficit grave de AAT (41mg/dl) siendo detectado el alelo PI*Z en heterocigosis. El estudio molecular del gen SERPINA1 reveló un genotipo Z/Q0cardiff (fig. 1). El paciente no mostraba síntomas respiratorios o de enfermedad hepática. Las pruebas funcionales respiratorias se encontraban dentro de límites normales: FEV1/FVC 0,79; FEV1 4,82 l (109%); FVC 5,60 l (112%); DLCO 109%; KCO 106%. La radiografía de tórax y la analítica general no mostraban datos relevantes.
La AAT es una antiproteasa producida principalmente por los hepatocitos, la cual se opone a la actividad de la elastasa de los neutrófilos y cuya concentración plasmática oscila entre 120-200mg/dl1. Si bien el alelo deficitario más frecuente y que conduce a niveles de ATT plásticos muy bajos (en torno al 10-15% del nivel normal) es la mutación Z (p.Glu342Lys), en las mutaciones denominadas nulas los niveles de AAT sérica son extremadamente bajos o indetectables3-9, encontrándose implicados en este desenlace una amplia gama de mecanismos moleculares entre los que se encuentran errores en la síntesis proteica o degradación postraducción6,10-12. Por ello, los genotipos constituidos por alelos nulos en homocigosis o acompañados de otros alelos deficientes del gen SERPINA1 conllevan un riesgo particularmente alto de presentar enfisema pulmonar de inicio muy temprano, incluso antes de lo que podría originarse en el genotipo ZZ13.
En lo referente al PI*Q0amersfoort la escasa literatura encontrada apunta a que, tanto las formas heterocigotas como homocigotas, desarrollan EPOC a una temprana edad de forma similar a nuestra paciente, sin describirse afectación hepática alguna7,8. Dicha mutación provoca un codón de parada en la posición 184 de la proteína dando lugar a un déficit grave al asociarse con otras variantes deficitarias como PI*Z. De forma similar a otros alelos nulos, la variante PI*Q0amersfoort no produce afectación hepática al no polimerizarse la proteína a nivel del hígado como ocurre en mutaciones por cambio de aminoácido, en donde existen defectos que alteran la estructura y el plegamiento proteico, dando lugar a su acumulación a nivel del retículo endoplasmático de los hepatocitos produciendo finalmente un daño tisular.
En el caso de PI*Q0cardiff se produce la sustitución del aminoácido aspartato por valina en posición 256 de la proteína ATT. Dicha sustitución produce un déficit grave en homocigosis o asociada a otras variantes deficitarias como puede ser PI*Z. Algunos autores defienden que los pacientes Q0cardiff en homocigosis no presentan riesgo alguno de enfisema, aunque podrían tenerlo en el caso de combinarse en heterocigosis con el alelo PI*Z u otros alelos nulos9,14. En lo que respecta a nuestro caso, el paciente se encontraba asintomático. En nuestra opinión, PI*Q0cardiff no puede ser considerado un alelo nulo, ya que no se trata de una mutación que provoque un codón de parada y produzca la degradación proteica completa, mecanismo habitual de los alelos deficitarios Null5. Algunos autores definen la variante PI*Q0cardiff como PI*P(lowell), consecuencia de que la variación genética consistente en el cambio de un ácido aspártico por valina9 provoca la degradación intracelular de la proteína, lo que conlleva a niveles reducidos de proteína aunque no indetectables15. Es probable que estos niveles residuales de proteína, junto con los producidos por el alelo PI*Z, hagan que nuestro paciente presente niveles de AAT por encima de los encontrados en pacientes portadores de alelos nulos.







