


Alpha-1 antitrypsin (AAT) is a medium-sized circulating glycoprotein encoded by the SERPINA1 gene, located on the short arm of chromosome 14, in the 14q32.13 region.1 AAT mainly acts as a protease inhibitor, and protects lung tissue from the proteolytic action of neutrophil elastase. Other characteristics, such as the inhibition of other serine proteases, and anti-inflammatory, immunomodulatory, and antimicrobial activity have also been described.2 AAT deficiency (AATD) is one of the most common genetic disorders in the Caucasian population. An AAT level below 80mg/dl, determined by nephelometry, increases the risk of pulmonary emphysema in adulthood,3 the most common form of disease associated with AATD. Other manifestations include various types of hepatitis in children and adults, panniculitis and vasculitis. An association with other inflammatory disease has been posited in recent years.
AAT exhibits simple, autosomal, codominant Mendelian inheritance. Around 85%–90% of the population has an MM genotype, formed from the inheritance of an allele from each parent, which synthesizes normal amounts of functioning protein. The most common deficient alleles are PI*S and Pi*Z, which occur in the Spanish population at an estimated prevalence of 10.4% and 1.72%, respectively, and express 40% and 15% of normal AAT levels, also respectively. In clinical practice, 96% of AATD disease occurs in ZZ homozygotes, and the remaining 4% occur in SZ and MZ heterozygotes and in the uncommon rare and null genotypes or their combinations with alleles PI*S and Pi*Z.
We describe the case of a family carrying a Q0Ourém gene,4 a null allele identified for the first time in the central region of Portugal, very few cases of which have been described in the literature.
A 2-year-old girl with a history of ferropenic anemia, mild gastroesophageal reflux, and mild-moderate persistent asthma with several hospital admissions due to exacerbations. She was receiving treatment with inhaled fluticasone, montelukast and salbutamol on demand. After an admission at the age of 11 months, she was investigated in the Pediatric Department for anorexia and growth retardation after the age of 8 months. Tests for anti-transglutaminase antibodies, immunoglobulins, skin prick tests for egg, fish, wheat and cow's milk protein (CMP), and viral serologies were negative. Thyroid function and chlorine in sweat were normal. AAT levels: 40mg/dl. Genetic study by sequencing of the SERPINA1 gene identified the PI*SQ0Ourém genotype.
The family study was completed with AAT determinations in the patient's parents. The father is a 33-year-old man, smoker of 10–15 cigarettes a day (pack-year index: 16.5), welder by profession. He had no history of lung disease nor was he receiving regular treatments. Physical examination was normal. Clinical laboratory tests were normal except for AAT 34mg/dl (116–232mg/dl/21–41μmol/l).5 The gene sequencing study identified genotype PI*SQ0Ourém. Lung function tests were normal. High-resolution computed tomography (HRCT) showed panlobular emphysema in anterior fields and bases with no signs of centrilobular emphysema. The patient is being monitored in the Respiratory Medicine outpatient clinic and has given up smoking. His lung function remains normal.
The mother was a 30-year-old woman with no significant medical history. AAT levels: 119mg/dl with PiMS phenotype.
The low number of cases with a null allele in patients with AATD (100–200 fold lower prevalence of allele PI*Z) limits the understanding and evaluation of the prevalence of these variants (Fig. 1). The null alleles give rise to a structurally dysfunctional protein, causing AAT to degrade within the cell, leading to undetectable levels in serum. For this reason, they are associated with a very high risk of emphysema but not of liver disease, due to the lack of polymerization.
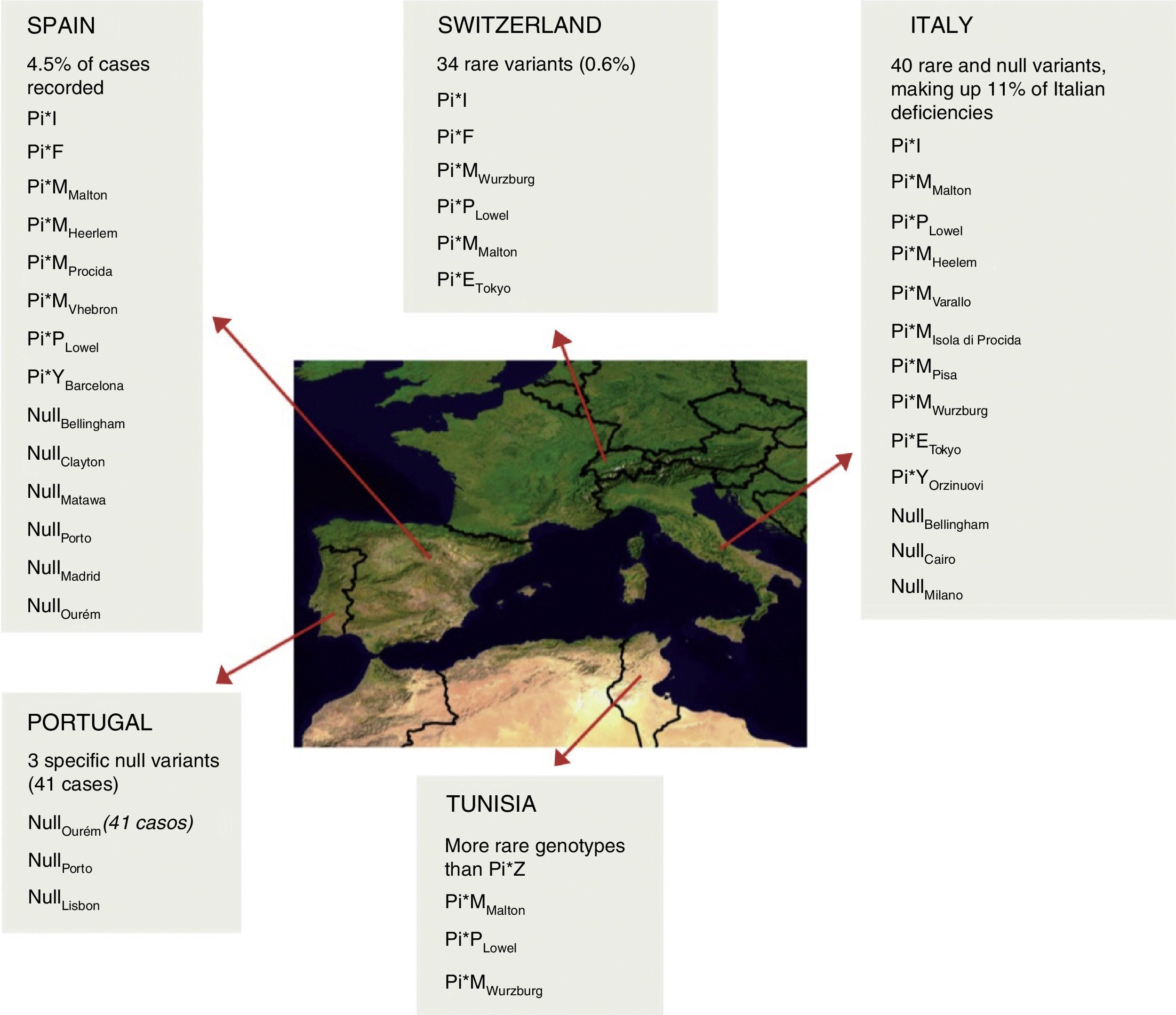
Distribution and estimated frequency of rare and null genotypes in the Iberian Peninsula, Switzerland, Italy and Tunisia.7
Our 2 patients are heterozygous carriers of an Q0Ourém allele, first described in 2002 in a very specific area in Portugal,4 and later in La Palma, Canary Islands, Spain.6 The Q0Ourém mutation is characterized by the insertion of a thymine nucleotide in the coding region of exon 5 within a small microsatellite (T)5, on a normal M3 genetic background. The resulting frameshift creates a premature stop codon that shortens the carboxy-terminal end of the 19-amino acid protein, that includes a proline residue essential for AAT secretion. The mutation, while not affecting the size or stability of the messenger RNA, causes the altered protein to be retained in the endoplasmic reticulum, where it is degraded.7 This mutation is identical to Q0Mattawa mutation, but the latter originates from an M1 genetic background.8 In a review of the historical evolution and clinical and functional impact of AAT deficiency caused by the Q0Ourém allele, published in 2012,9 41 individuals carriers of this allele were identified among 4 families in the central area of Portugal. Of these, 8 were homozygous, 5 had PI*SQ0Ourém and 28 had PI*MQ0Ourém. None of the 5 individuals with PI*SQ0Ourém were smokers and all patients had normal lung function and chest X-ray results. HRCT was not performed in any of these cases. In our adult patient, the finding of emphysema in the HRCT at the age of 33 years may be associated with the high susceptibility of individuals with severe AAT deficiency to the effects of smoking. This underlines the importance of an early diagnosis of AADT, in order to avoid the patient starting smoking, or to encourage cessation in those who already smoke.
Five individual carriers of the PI*Q0Ourém allele were identified in La Palma. The index case had PI*SQ0Ourém, 3 had PI*MQ0Ourém and 1 had PI*ZQ0Ourém.
This mutation is estimated to be comparatively recent, having emerged 650 years ago, in the 14th century. The origin of the variant has been established as the center of Portugal. The geographical proximity of this area with Galicia would explain the presence of this allele among the population of Galicia, but to our knowledge, these are the 2 first cases of this mutation published in Galicia, in 2 members of the same family.
Please cite this article as: Tubío-Pérez RA, Blanco-Pérez M, Ramos-Hernández C, Torres-Durán M. Descripción de la deficiencia de alfa-1-antitripsina asociada al alelo PI*Q0Ourém en una niña de 2 años de edad y su estudio familiar. Un caso infrecuente. Arch Bronconeumol. 2018;54:228–230.







