





Understanding the diverse pathogenetic pathways in obstructive sleep apnea (OSA) is crucial for improving outcomes. microRNA (miRNA) profiling is a promising strategy for elucidating these mechanisms.
ObjectiveTo characterize the pathogenetic pathways linked to OSA through the integration of miRNA profiles, machine learning (ML) and bioinformatics.
MethodsThis multicenter study involved 525 patients with suspected OSA who underwent polysomnography. Plasma miRNAs were quantified via RNA sequencing in the discovery phase, with validation in two subsequent phases using RT-qPCR. Supervised ML feature selection methods and comprehensive bioinformatic analyses were employed. The associations among miRNA targets, OSA and OSA treatment were further explored using publicly available external datasets.
ResultsFollowing the discovery and technical validation phases in a subset of patients with and without confirmed OSA (n=53), eleven miRNAs were identified as candidates for the subsequent feature selection process. These miRNAs were then quantified in the remaining population (n=472). Feature selection methods revealed that the miRNAs let-7d-5p, miR-15a-5p and miR-107 were the most informative of OSA. The predominant mechanisms linked to these miRNAs were closely related to cellular events such as cell death, cell differentiation, extracellular remodeling, autophagy and metabolism. One target of let-7d-5p and miR-15a-5p, the TFDP2 gene, exhibited significant differences in gene expression between subjects with and without OSA across three independent databases.
ConclusionOur study identified three plasma miRNAs that, in conjunction with their target genes, provide new insights into OSA pathogenesis and reveal novel regulators and potential drug targets.

Obstructive sleep apnea (OSA) is a prevalent sleep disorder characterized by the recurrent occurrence of obstructive apnea and hypopnea, leading to repeated decreases in oxygen saturation and disrupted sleep patterns. OSA is an important contributor to poor health outcomes, including a spectrum of morbidities and an elevated risk of major causes of mortality.1,2 OSA is a complex condition with a heterogeneous profile of clinical and physiological phenotypes,3,4 presenting a challenge in tailoring effective therapeutic strategies. The identification of the pathogenetic pathways is a pivotal step in improving outcomes in OSA patients. Continuous positive airway pressure (CPAP) represents the cornerstone therapeutic intervention. Despite its well-established efficacy in reducing daytime somnolence and enhancing quality of life, a proportion of individuals treated with CPAP continues to experience residual symptoms and associated comorbidities.5 This scenario suggests the necessity for adjuvant therapies capable of complementing CPAP therapy and addressing the multifactorial nature of OSA.
microRNAs (miRNAs) are small noncoding RNAs (ncRNAs) composed of 19–25 nucleotides that play a key role in the posttranscriptional regulation of gene expression. miRNAs typically inhibit gene expression by binding to target mRNAs, thereby inducing their degradation or translational repression. However, in specific instances, miRNAs can also activate translation.6 It has been predicted that a significant proportion of the human transcriptome is regulated by miRNAs.7 As such, miRNAs are mediators of numerous fundamental biological processes and play significant roles in various diseases, including respiratory conditions.8 Previous studies have shown that miRNAs play a role in various pathobiological mechanisms associated with OSA. For example, the SREBP2/miR-210 axis and its impact on mitochondrial dysfunction represent a mechanistic link between OSA and endothelial cell dysfunction.9 The downregulation of miR-15b-5p and miR-92b-3p in OSA patients may contribute to intermittent hypoxia/reoxygenation-induced oxidative stress by influencing the eicosanoid inflammatory pathway.10
Over the past decade, miRNAs have emerged as crucial tools for developing next-generation therapeutics. Numerous well-designed studies have demonstrated the efficacy and safety of miRNA-based treatments.11,12 A notable example is miravirsen, a locked nucleic acid–modified DNA phosphorothioate antisense oligonucleotide that sequesters mature miR-122, resulting in a dose-dependent and sustained reduction in hepatitis C virus (HCV) RNA levels in patients with chronic HCV genotype 1 infection.13 In addition, second-line therapies using miRNA mimics, such as miR-34a and let-7g, are being investigated to reduce tumor burden and promote tumor suppression.14,15 Beyond their intracellular location, miRNAs serve as mediators of intercellular communication through their secretion into the extracellular milieu and regulation of gene expression in recipient cells.16–18 Although the hormone-like effect of miRNAs has been a topic of debate,19 a multitude of studies have provided evidence supporting their role as endocrine genetic signals.20–22 Overall, the circulating miRNome has emerged as a promising tool for improving the mechanistic understanding of disease and identifying novel therapeutic treatments.
The analysis of ncRNAs, including miRNAs, constitutes a valuable strategy in the field of network medicine, as it facilitates the discovery of novel mechanistic insights that were previously elusive.23,24 Investigating these transcripts enhances the ability to identify molecular mechanisms associated with a given disease. Notably, the integration of extracellular ncRNA profiling with machine learning (ML) methods has been previously employed to identify molecular drivers of disease.25 The distinct advantage of ML is its ability to decipher previously unknown multidimensional interactions between predictors and outcomes and therefore to identify novel biological features. This is particularly relevant for miRNAs, given their involvement in regulating gene expression through dynamic, complex and coordinated networks.26
In the current investigation, we synergistically employed circulating cell-free miRNome profiling with ML feature selection techniques and bioinformatic analyses. This integrative approach aimed to characterize the mechanistic pathways associated with OSA and to propose innovative disease-modifying agents.
MethodsMethods for blood collection, miRNA sequencing and external dataset are provided in Supplemental Material.
Study PopulationThe study included 525 subjects referred to the sleep unit due to suspected OSA (ClinicalTrials.gov identifier: NCT03513926). The recruitment was conducted at University Hospital Arnau de Vilanova-Santa María of Lleida and Hospital San Pedro Alcántara of Cáceres in Spain. Patients aged older than 18 years were recruited for the present study. The exclusion criteria were the presence of a previously diagnosed sleep disorder, a history of CPAP treatment, inability to complete the questionnaires and any medical, social or geographic factors that could compromise the eligibility of the subject, i.e., pregnancy, substance abuse, cancer, renal insufficiency, severe chronic obstructive pulmonary disease, chronic depression and other very limiting chronic diseases or life expectancy less than 1 year. General physical and anthropometric parameters were documented, and information on sociodemographic characteristics, medical history, medication use and lifestyle habits was collected by trained clinicians.
Sleep EvaluationAll eligible participants underwent a comprehensive polysomnographic (PSG) sleep study (Sleepware G3, Philips, Amsterdam, Netherlands). All procedures were conducted according to national guidelines and regulations governing clinical practice.27 Trained personnel who adhered to standard criteria28 analyzed the results of the sleep studies. Apnea was characterized as an interruption or reduction in oronasal airflow by ≥90% for at least 10s, while hypopnea was defined as a 30–90% reduction in oronasal airflow for at least 10s, associated with either oxygen desaturation by at least 3% or an arousal on the electroencephalogram. The apnea–hypopnea index (AHI), which is indicative of OSA severity, was calculated based on the average number of apnea and hypopnea events per hour of sleep. Following the PSG study, subjects were categorized according to the International Consensus Document on Obstructive Sleep Apnea29 into non-OSA (AHI<15events/h) and OSA (AHI≥15events/h) groups.
EthicsAll subjects provided written informed consent for study participation prior to the collection of blood samples. This study was revised and approved by the Clinical Research Ethics Committee of the University Hospital Arnau de Vilanova-Santa María of Lleida (no. 1153/1411). The study was performed according to the Declaration of Helsinki.
Study DesignThe study design is presented in Fig. 1. The experimental approach was composed of three clearly defined phases: (i) Discovery phase: from the initial population of 525 participants with suspected OSA, 27 subjects with confirmed OSA were randomly selected. Subsequently, 26 controls without OSA from the same study population were selected using nearest neighbor propensity score matching by age, sex and body mass index (BMI). miRNA sequencing was conducted on these 53 subjects. The number of samples aligns with the recommendations for transcriptomic profiling studies.30 (ii) Technical validation phase: to confirm the reliability of the miRNA sequencing findings, a rigorous assessment of miRNA candidates was conducted using RT-qPCR, which is acknowledged as the gold standard for circulating miRNA quantification. This technical validation was performed on the same set of samples employed in the discovery phase (n=53). (iii) Comprehensive evaluation: miRNA candidates that successfully passed the technical validation phase were further evaluated in the entire study population, excluding samples utilized in the previous steps (n=472).
Plasma microRNA Profiling, license number: TH26UP0M73. Abbreviations: CPAP: continuous positive airway pressure; OSA: obstructive sleep apnea.")
Total RNA was extracted from 200μL of frozen plasma utilizing the miRNeasy Serum/Plasma Advanced Kit (Qiagen, Hilden, Germany). To establish a normalization strategy, synthetic Caenorhabditis elegans miR-39-3p (cel-miR-39-3p) (Qiagen) was introduced as an external reference miRNA at a concentration of 1.6×108copies/μL. Additionally, 1μg of MS2 carrier RNA (Roche Diagnostics, Mannheim, Germany) was added to the mixture to increase the yield of extracellular miRNAs. Quality control for RNA isolation was assured by the inclusion of the RNA Spike-In Kit (synthetic UniSp2, UniSp4, and UniSp5) (Qiagen). All reagents were spiked into samples during RNA isolation after incubation with the denaturing solution. The isolated RNA was then eluted in 20μL of RNAse-free water and stored at −80°C.
For miRNA quantification, the miRCURY LNA Universal RT microRNA PCR System (Qiagen) protocol was followed. Reverse transcription (RT) to synthesize complementary DNA (cDNA) was performed using a miRCURY LNA RT Kit (Qiagen) in a total reaction volume of 10μL. The spike-in UniSp6 (Qiagen) was added to monitor the RT reaction. The RT conditions included incubation at 42°C for 60min, inactivation at 95°C for 5min and immediate cooling at 4°C. Subsequently, the cDNA was stored at −20°C. We used the miRCURY LNA miRNA Custom Panels (384-well plates) (Qiagen), which contained the selected miRNAs and spike-in primers (Qiagen). A QuantStudio™ 7 Flex Real-Time PCR System (Applied Biosystems, Waltham, MA, USA) was used for qPCR in a 10μL reaction volume. The RT-qPCR conditions included an initial incubation at 95°C for 2min, followed by 40 cycles of 95°C for 10s and 56°C for 1min and a final melting curve analysis. Synthetic UniSp3 served as an interplate calibrator and qPCR control. Amplification curves were assessed by melting curve analysis using QuantStudio Software v1.3 (Thermo Fisher Scientific, MA, USA), ensuring the presence of single products and the absence of primer dimers.
The quantification cycle (Cq) was determined as the fractional cycle number at which the fluorescence surpassed a defined threshold. Cq values of spike-in RNA templates were initially scrutinized to monitor the homogenous efficiencies of RNA extraction procedures, the robustness of RT and the absence of PCR inhibitors. The ΔCq ratio (miR-23a-3p–miR-451a), as proposed by Blondal et al.,31 was used to exclude hemolyzed samples. Cq values exceeding 35 cycles were considered undetectable and censored at the minimum level observed for each miRNA. miRNAs detected below the limit of detection in more than 80% of the samples were categorized as nonexpressed. Samples that did not pass the quality control test were excluded from subsequent statistical analysis. Relative quantification was accomplished using the 2−Cq method (ΔCq=CqmiRNA−Cqcel-miR-39-3p). The expression levels were log-transformed for subsequent statistical analysis.
Functional AssessmentWe retained the predicted miRNA–target interactions utilizing the web-based TargetScan (Release 8.0, https://www.targetscan.org/vert_80/).32 The search settings were set to all predicted targets, regardless of conservation. To elucidate the biological relevance and pathways associated with the identified miRNA targets, we conducted a functional enrichment analysis using the R package ClusterProfiler 4.6.2.33ClusterProfiler is released through the Bioconductor project and can be accessed via https://bioconductor.org/packages/clusterProfiler/. The Kyoto Encyclopedia of Genes and Genomes (KEGG), Gene Ontology (GO) and Reactome were used as reference databases. To further explore the expression patterns of selected miRNA targets, tissue patterns from the Genotype-Tissue Expression (GTEx) Portal (https://www.gtexportal.org/home/) project v.8 were used. To explore potential therapeutic interventions, we investigated the repositioning potential of upregulated miRNA targets using the Drug–Gene Interaction database (DGIdb v.5.0.5) (https://dgidb.org).34
Statistical AnalysisAll the statistical analyses were performed using R version 4.2.2 (R Foundation for Statistical Computing). Descriptive statistics were employed to explore the characteristics of the study population. Continuous variables were compared between groups using the Mann–Whitney U test, whereas categorical variables were compared using Fisher's exact test. The data are presented as frequencies (percentages) for categorical variables and as medians (25th and 75th percentiles) for continuous variables. Spearman correlation was utilized to estimate the correlation between continuous variables. A correlation coefficient (rho) greater than 0.3 was considered as biologically relevant.35 The p-value threshold defining statistical significance was set at <0.05.
Three distinct ML-based feature selection methods were implemented simultaneously: Random forest with Boruta,36 variable selection using random forests (VSURF)37 and sparse partial least squares (sPLS).38 To ensure the robustness and consistency of our selection process, we repeated each method 50 times. In each iteration, miRNAs selected in at least one run were considered important variables. Finally, miRNAs consistently identified across all three methods were designated as candidates for further investigation through bioinformatics analysis, as previously described.39
For the external datasets, we obtained the preprocessed gene expression data and normalized them using quantile normalization. The raw data were processed and normalized using methods implemented in the Limma R package40 for the Illumina and Agilent microarrays and the oligo R package41 for the Affymetrix chips. Subsequently, we applied a base 2 logarithmic scale transformation. Following data normalization, we averaged the expression data from multiple probes associated with the same gene. An extensive outlier analysis was performed to assess the plausibility of extreme values. After thorough evaluation, outliers were excluded from further analysis. Differential expression analysis was performed with processed data between groups of each microarray dataset using the Limma R package. For the longitudinal analysis, we used a paired-samples design to account for interindividual differences with respect to the pretreatment state. For each comparison, we utilized the Limma moderated t-test (implemented with the “lmFit” and “eBayes” functions) to calculate differential expression for each gene.
ResultsSelection of microRNA CandidatesmiRNA sequencing was conducted in specific matched subgroups of subjects with and without confirmed OSA (n=53). The main characteristics of the study groups are detailed in Table 1. miRNA candidates were chosen based on the following criteria: over the 90th decile of expression (to ensure their detection in the following phases), ≥1.3-fold differential expression and p-value≤0.2 (to identify a broader range of potential biomarkers). A total of 38 miRNAs fulfilled these criteria, as depicted in Fig. 2A.
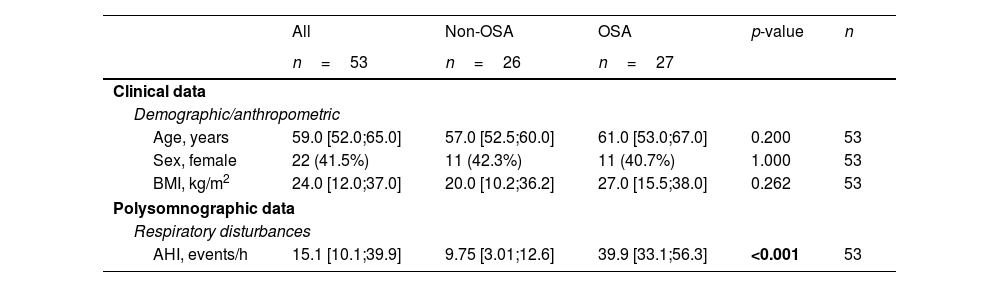
Clinical Characteristics of the Study Population at Baseline (Discovery and Technical Validation Phases).
| All | Non-OSA | OSA | p-value | n | |
|---|---|---|---|---|---|
| n=53 | n=26 | n=27 | |||
| Clinical data | |||||
| Demographic/anthropometric | |||||
| Age, years | 59.0 [52.0;65.0] | 57.0 [52.5;60.0] | 61.0 [53.0;67.0] | 0.200 | 53 |
| Sex, female | 22 (41.5%) | 11 (42.3%) | 11 (40.7%) | 1.000 | 53 |
| BMI, kg/m2 | 24.0 [12.0;37.0] | 20.0 [10.2;36.2] | 27.0 [15.5;38.0] | 0.262 | 53 |
| Polysomnographic data | |||||
| Respiratory disturbances | |||||
| AHI, events/h | 15.1 [10.1;39.9] | 9.75 [3.01;12.6] | 39.9 [33.1;56.3] | <0.001 | 53 |
Data are presented as the median [25th;75th percentile] for continuous variables and as frequencies (percentage) for categorical variables. p-Values<0.05 are presented in bold. AHI=apnea–hypopnea index; BMI=body mass index; OSA=obstructive sleep apnea.
 Volcano plot showing the p-value versus the fold change for the microRNAs analyzed in the discovery phase. microRNAs over the 90th decile of expression are depicted in dark gray. The green dots indicate microRNA candidates. (B) Violin plots including microRNAs that fulfilled the selection criteria in the technical validation phase. Plots depict log102−ΔCq as the microRNA expression unit. The gray color indicates the nonobstructive sleep apnea group, and the red color indicates the obstructive sleep apnea group. The plot presents the median (25th and 75th percentiles) estimator of the density (as density curves) and individual values (black dots). Fold changes and p-values are displayed. (C) Correlogram showing spearman correlation coefficients between polysomnographic parameters and the microRNAs that fulfilled the selection criteria in the technical validation phase. The color scale illustrates the degree of correlation and ranges, indicating the negative to positive correlations. Abbreviations: AHI: apnea–hypopnea index; OSA: obstructive sleep apnea; SaO2: oxygen saturation; TSat90: time during the night with SaO2 below 90%.")
Identification of microRNAs related to obstructive sleep apnea. (A) Volcano plot showing the p-value versus the fold change for the microRNAs analyzed in the discovery phase. microRNAs over the 90th decile of expression are depicted in dark gray. The green dots indicate microRNA candidates. (B) Violin plots including microRNAs that fulfilled the selection criteria in the technical validation phase. Plots depict log102−ΔCq as the microRNA expression unit. The gray color indicates the nonobstructive sleep apnea group, and the red color indicates the obstructive sleep apnea group. The plot presents the median (25th and 75th percentiles) estimator of the density (as density curves) and individual values (black dots). Fold changes and p-values are displayed. (C) Correlogram showing spearman correlation coefficients between polysomnographic parameters and the microRNAs that fulfilled the selection criteria in the technical validation phase. The color scale illustrates the degree of correlation and ranges, indicating the negative to positive correlations. Abbreviations: AHI: apnea–hypopnea index; OSA: obstructive sleep apnea; SaO2: oxygen saturation; TSat90: time during the night with SaO2 below 90%.
To validate the miRNA sequencing results, the selected candidates were further examined through RT-qPCR using the same sample set (n=53). Eight samples were excluded from further analysis due to failing the quality control test, which was attributed to the presence of hemolysis and/or low quality of the spike-ins. miR-6877-5p was expressed at low levels (Cq≥35 in more than 80% of the samples) and was therefore excluded from further analysis. Among the remaining 37 miRNA candidates, those with a ≥1.3-fold difference between study groups, with a p-value≤0.01 and that correlated with the AHI (rho>0.3) were retained (Fig. 2B, C and Supplemental Table S1). Ultimately, 11 miRNAs—let-7d-5p, miR-15a-5p, miR-15b-5p, miR-18a-5p, miR-20b-5p, miR-30e-5p, miR-107, miR-199a-5p, miR-425-5p, miR-484 and miR-625-5p—were identified as candidates for the subsequent feature selection process. With the exception of the AHI and the apnea index, weak correlations were observed between the miRNA panel and the parameters obtained from PSG (Fig. 2C).
Feature Selection Procedure and BioinformaticsFollowing the identification of miRNA candidates associated with OSA, quantification was performed using RT-qPCR in the entire study population. A total of 9.5% of the samples did not pass quality control due to hemolysis or variability in spike-ins and were discarded from further analysis. The detailed characteristics of the study population are presented in Table 2. As anticipated, patients with OSA were older, predominantly men, had a higher BMI and had a more unfavorable profile of comorbidities and disease-related sleep parameters.
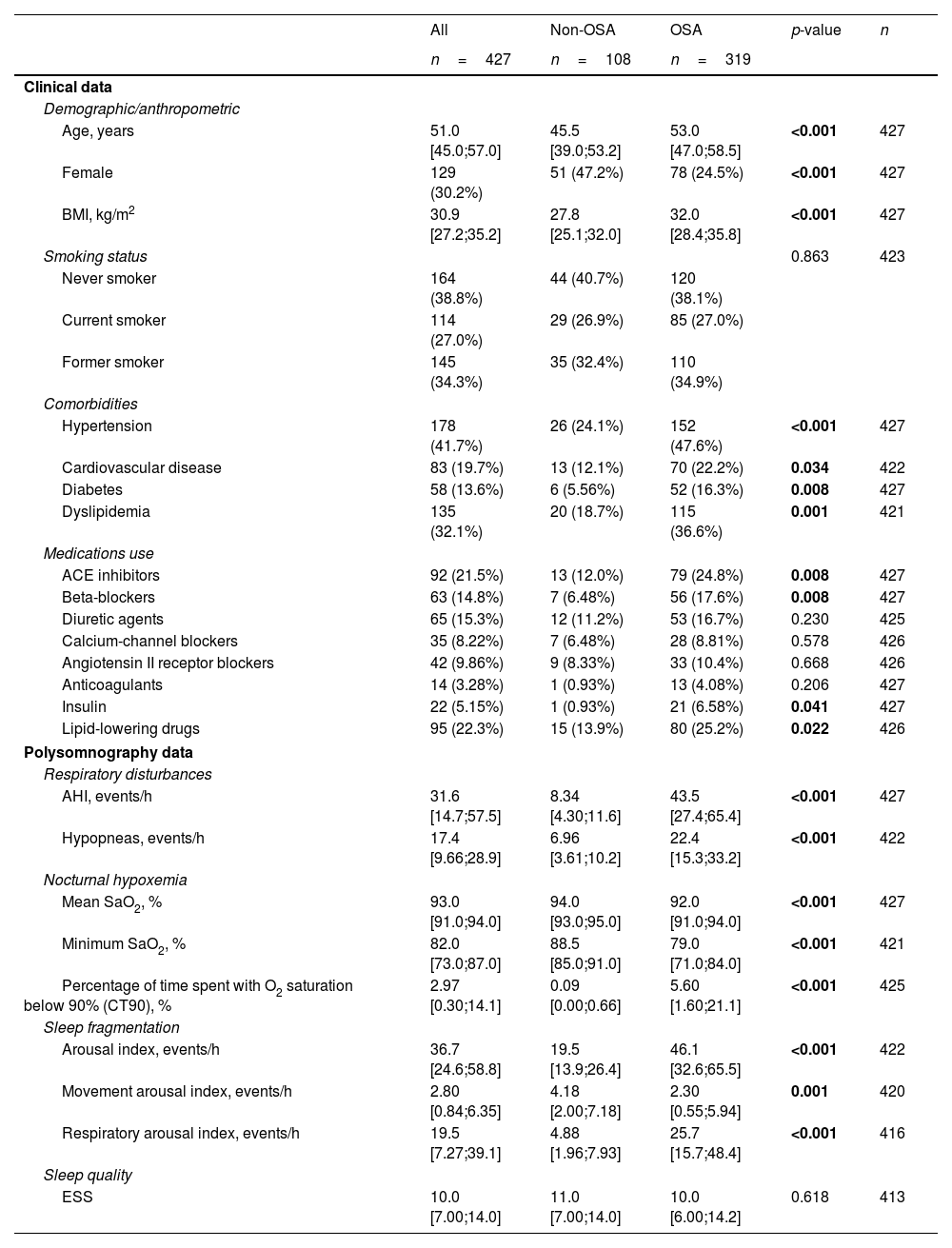
Characteristics of the Study Population (Comprehensive Evaluation).
| All | Non-OSA | OSA | p-value | n | |
|---|---|---|---|---|---|
| n=427 | n=108 | n=319 | |||
| Clinical data | |||||
| Demographic/anthropometric | |||||
| Age, years | 51.0 [45.0;57.0] | 45.5 [39.0;53.2] | 53.0 [47.0;58.5] | <0.001 | 427 |
| Female | 129 (30.2%) | 51 (47.2%) | 78 (24.5%) | <0.001 | 427 |
| BMI, kg/m2 | 30.9 [27.2;35.2] | 27.8 [25.1;32.0] | 32.0 [28.4;35.8] | <0.001 | 427 |
| Smoking status | 0.863 | 423 | |||
| Never smoker | 164 (38.8%) | 44 (40.7%) | 120 (38.1%) | ||
| Current smoker | 114 (27.0%) | 29 (26.9%) | 85 (27.0%) | ||
| Former smoker | 145 (34.3%) | 35 (32.4%) | 110 (34.9%) | ||
| Comorbidities | |||||
| Hypertension | 178 (41.7%) | 26 (24.1%) | 152 (47.6%) | <0.001 | 427 |
| Cardiovascular disease | 83 (19.7%) | 13 (12.1%) | 70 (22.2%) | 0.034 | 422 |
| Diabetes | 58 (13.6%) | 6 (5.56%) | 52 (16.3%) | 0.008 | 427 |
| Dyslipidemia | 135 (32.1%) | 20 (18.7%) | 115 (36.6%) | 0.001 | 421 |
| Medications use | |||||
| ACE inhibitors | 92 (21.5%) | 13 (12.0%) | 79 (24.8%) | 0.008 | 427 |
| Beta-blockers | 63 (14.8%) | 7 (6.48%) | 56 (17.6%) | 0.008 | 427 |
| Diuretic agents | 65 (15.3%) | 12 (11.2%) | 53 (16.7%) | 0.230 | 425 |
| Calcium-channel blockers | 35 (8.22%) | 7 (6.48%) | 28 (8.81%) | 0.578 | 426 |
| Angiotensin II receptor blockers | 42 (9.86%) | 9 (8.33%) | 33 (10.4%) | 0.668 | 426 |
| Anticoagulants | 14 (3.28%) | 1 (0.93%) | 13 (4.08%) | 0.206 | 427 |
| Insulin | 22 (5.15%) | 1 (0.93%) | 21 (6.58%) | 0.041 | 427 |
| Lipid-lowering drugs | 95 (22.3%) | 15 (13.9%) | 80 (25.2%) | 0.022 | 426 |
| Polysomnography data | |||||
| Respiratory disturbances | |||||
| AHI, events/h | 31.6 [14.7;57.5] | 8.34 [4.30;11.6] | 43.5 [27.4;65.4] | <0.001 | 427 |
| Hypopneas, events/h | 17.4 [9.66;28.9] | 6.96 [3.61;10.2] | 22.4 [15.3;33.2] | <0.001 | 422 |
| Nocturnal hypoxemia | |||||
| Mean SaO2, % | 93.0 [91.0;94.0] | 94.0 [93.0;95.0] | 92.0 [91.0;94.0] | <0.001 | 427 |
| Minimum SaO2, % | 82.0 [73.0;87.0] | 88.5 [85.0;91.0] | 79.0 [71.0;84.0] | <0.001 | 421 |
| Percentage of time spent with O2 saturation below 90% (CT90), % | 2.97 [0.30;14.1] | 0.09 [0.00;0.66] | 5.60 [1.60;21.1] | <0.001 | 425 |
| Sleep fragmentation | |||||
| Arousal index, events/h | 36.7 [24.6;58.8] | 19.5 [13.9;26.4] | 46.1 [32.6;65.5] | <0.001 | 422 |
| Movement arousal index, events/h | 2.80 [0.84;6.35] | 4.18 [2.00;7.18] | 2.30 [0.55;5.94] | 0.001 | 420 |
| Respiratory arousal index, events/h | 19.5 [7.27;39.1] | 4.88 [1.96;7.93] | 25.7 [15.7;48.4] | <0.001 | 416 |
| Sleep quality | |||||
| ESS | 10.0 [7.00;14.0] | 11.0 [7.00;14.0] | 10.0 [6.00;14.2] | 0.618 | 413 |
Data are presented as the median [25th;75th percentile] for quantitative variables and as frequencies (percentage) for qualitative variables. p-Values<0.05 are presented in bold. Definitions of abbreviations: ACE=angiotensin-converting enzyme; AHI=apnea–hypopnea index; BMI=body mass index; ESS=Epworth sleepiness scale; OSA=obstructive sleep apnea; SaO2=oxygen saturation.
Due to the intricate dynamics of miRNA networks, the circulating levels of miRNAs were entered into a supervised feature selection protocol that integrates three distinct ML-based methods (Fig. 3A). The outcome of each method resulted in a unique feature subset: (i) VSURF selected eight candidates: let-7d-5p, miR-15a-5p, miR-18a-5p, miR-30e-5p, miR-107, miR-199a-5p, miR-425-5p and miR-484; (ii) sPLS selected six candidates: let-7d-5p, miR-15a-5p, miR-15b-5p, miR-20b-5p, miR-107 and miR-625-5p; and (iii) Boruta selected all candidates. Notably, let-7d-5p, miR-15a-5p and miR-107 demonstrated robust and consistent selection across the three methods.
 Identification of microRNAs associated with obstructive sleep apnea using a consensus of three supervised machine learning feature selection algorithms (Boruta, VSURF and sPLS) with the intersected microRNAs among the methods selected as candidates. (B–D) Functional enrichment analysis of the predicted downstream target genes using KEGG (B), GO (C) and Reactome (D) in the R package ClusterProfileR 4.0. The plots present the rich factors of the pathways with the most significant differences, considering their p-values. The intensity of the colors of the bars denotes the p-values. The rich factor consists of a ratio of target genes annotated in the molecular processes to all genes annotated in the processes.")
Identification of microRNA-mediated molecular mechanisms associated with obstructive sleep apnea. (A) Identification of microRNAs associated with obstructive sleep apnea using a consensus of three supervised machine learning feature selection algorithms (Boruta, VSURF and sPLS) with the intersected microRNAs among the methods selected as candidates. (B–D) Functional enrichment analysis of the predicted downstream target genes using KEGG (B), GO (C) and Reactome (D) in the R package ClusterProfileR 4.0. The plots present the rich factors of the pathways with the most significant differences, considering their p-values. The intensity of the colors of the bars denotes the p-values. The rich factor consists of a ratio of target genes annotated in the molecular processes to all genes annotated in the processes.
Target prediction for let-7d-5p, miR-15a-5p and miR-107 utilizing TargetScan v8.0 revealed a total of 3436 transcripts as potential targets (1993 targets for let-7d-5p, 1605 targets for miR-15a-5p and 101 targets for miR-107), with 257 targets shared by at least two of the selected miRNAs. To elucidate the underlying molecular pathways and biological processes associated with these identified target transcripts, an exhaustive enrichment analysis was conducted employing the KEGG, GO and Reactome databases. The analyses identified 9 KEGG pathways, 220 GO processes and 15 Reactome pathways (Fig. 3B–D and Supplemental Tables S2–S4). The predominant significant pathways and processes were closely linked to cellular events such as cell death, cell differentiation, extracellular remodeling, autophagy and metabolism.
External DatasetsTo assess the robustness of our findings, we systematically validated the results by analyzing the expression of the target transcripts in external datasets obtained from GEO. This process was divided into two steps. Initially, we conducted a comparative examination of miRNA targets between individuals with and without OSA. Subsequently, we analyzed the influence of CPAP therapy on the identified miRNA targets.
First, three distinct external datasets, including OSA patients and non-OSA subjects, were evaluated (GSE226379, GSE135917 and GSE75097). Among the targetomes of let-7d-5p, miR-15a-5p and miR-107, one gene, the transcription factor Dp-2 (TFDP2), which is a target of let-7d-5p and miR-15a-5p, exhibited significant differential expression in the three comparisons (fold change≥1.5 and p-value≤0.05) (Fig. 4A–C). To further elucidate the expression profile of the miRNA targets, we retrieved data from the GTEx project, which revealed that TFDP2 is expressed in most human tissues (Fig. 4D).
 Volcano plots showing the p-value versus the fold change for the predicted targetome in three external RNA sequencing datasets obtained from the GEO database from patients with obstructive sleep apnea. Significantly differentially expressed genes are presented in green (fold change>1.5 and p-value≤0.05). Intersected genes among the datasets are labeled. (A) Plasma, GSE226379; (B) subcutaneous fat, GSE135917; and (C) peripheral blood mononuclear cells (PBMCs), GSE75097. (D) Tissue enrichment analysis using GTEx (https://www.gtexportal.org/home/). Each column denotes a tissue type, and the row represents the intersected gene among the external datasets. The size and color of the points reflect the expression level of the gene, represented as normalized transcripts per million (nTPM) values. The GTEx Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health and by the NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. (E, F) Volcano plots showing the p-value versus the fold change for the predicted targetome in two external RNA sequencing datasets, including obstructive sleep apnea patients allocated to CPAP treatment, obtained from the GEO database. Intersected genes among the A–C datasets are labeled. (E) PBMCs, GSE133601; and (F) peripheral blood leukocytes, GSE49800.")
External datasets. (A–C) Volcano plots showing the p-value versus the fold change for the predicted targetome in three external RNA sequencing datasets obtained from the GEO database from patients with obstructive sleep apnea. Significantly differentially expressed genes are presented in green (fold change>1.5 and p-value≤0.05). Intersected genes among the datasets are labeled. (A) Plasma, GSE226379; (B) subcutaneous fat, GSE135917; and (C) peripheral blood mononuclear cells (PBMCs), GSE75097. (D) Tissue enrichment analysis using GTEx (https://www.gtexportal.org/home/). Each column denotes a tissue type, and the row represents the intersected gene among the external datasets. The size and color of the points reflect the expression level of the gene, represented as normalized transcripts per million (nTPM) values. The GTEx Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health and by the NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. (E, F) Volcano plots showing the p-value versus the fold change for the predicted targetome in two external RNA sequencing datasets, including obstructive sleep apnea patients allocated to CPAP treatment, obtained from the GEO database. Intersected genes among the A–C datasets are labeled. (E) PBMCs, GSE133601; and (F) peripheral blood leukocytes, GSE49800.
To explore the impact of the current therapeutic strategy, the gene expression levels of TFDP2 were evaluated in two independent external datasets (GSE133601 and GSE49800), including samples from patients with OSA collected before and after CPAP therapy. No alterations in the transcript levels were observed following therapy (fold change≥1.5 and p-value≤0.05) (Fig. 4E and F). Next, we evaluated the repositioning potential of the miRNA target using DGIdb. No FDA-approved drugs were identified.
DiscussionRecognizing and characterizing the diverse pathogenetic pathways of OSA are pivotal steps toward improving patient outcomes. In this study, we implemented a complete approach that synergistically integrates plasma miRNA profiling with ML feature selection techniques and bioinformatic analyses to elucidate the mechanistic pathways associated with OSA and to propose innovative disease-modifying agents.
Through the combination of a high-throughput technique, i.e., sequencing, and the gold-standard methodology, i.e., RT-qPCR, we identified a set of 11 plasma miRNAs potentially linked to OSA. miRNAs play a significant role in finely regulating gene expression through their multifaceted interactions. A comprehensive understanding of the dynamics of miRNA-mediated regulation is crucial for deciphering their impact on cellular function and molecular pathways. ML methodologies capture complex and nonlinear relationships that may remain elusive using conventional univariable analyses.42 Consequently, the candidates quantified in the whole population were subjected to a ML feature selection process to define the most informative miRNAs. A combination of three distinct feature selection methods was employed to limit the number of selected miRNAs, thereby enhancing confidence and reproducibility. Our approach revealed three relevant miRNAs: let-7d-5p, miR-15a-5p and miR-107. The use of ML-based feature selection algorithms represents an innovative approach in OSA research, limiting direct comparisons with previous findings obtained using classical statistical approaches.43 Nevertheless, our candidates have been previously associated with OSA. Serum levels of miR-107 have been shown to be significantly altered in OSA patients compared with non-OSA controls.44 More recently, a study including individuals referred to the otorhinolaryngology service due to snoring or apnea revealed that both let-7d-5p and miR-107 were differentially expressed in nonhypertensive OSA and hypertensive OSA patients compared with controls, respectively.45 Furthermore, Santamaria-Martos et al.46 demonstrated the association of miR-107 with OSA severity parameters such as the AHI and arousal index. Consequently, earlier investigations provide evidence that the miRNAs identified here may be associated with OSA, further validating our ML approach.
In the next step, we used predicted miRNA interactions and enrichment analyses to provide novel insights into OSA pathobiology. Our analysis defined an array of specific mechanisms associated with the disease. Using three independent public datasets generated from different tissues, i.e., plasma, peripheral blood mononuclear cells (PBMCs) and subcutaneous fat, our approach identified TFDP2 as a potential contributor to OSA pathology. TFDP2, a member of the DP transcription factor family, forms heterodimers with E2F transcription factors, facilitating the transcriptional activation of genes critical for cell proliferation, differentiation and programmed cell death. For example, loss of TFDP2 disrupts the normal downregulation of E2F2 target genes, which are essential for regulating the cell cycle. This disruption leads to an accumulation of cells in the S phase and an increase in erythrocyte size.47 Importantly, prior research has established TFDP2 as a target of specific miRNAs. Bone marrow mesenchymal stem cells alleviate renal injury and fibrosis by modulating the miR-146a-5p/TFDP2 axis in mouse renal tubular epithelial cells.48 Furthermore, E2F transcription factors have been recognized as critical mediators of apoptosis across multiple experimental models and conditions.49,50 Although no existing literature directly associates TFDP2 with pathogenic mechanisms in OSA, the biological role of this mediator may explain our findings. Apoptosis, a cellular process essential for maintaining cellular homeostasis and survival, is a well-described pathogenetic mechanism in the context of OSA. Intermittent hypoxia, a hallmark of OSA, has been implicated in the generation of reactive oxygen species (ROS), which trigger endoplasmic reticulum stress and inhibit the synthesis of functional proteins, ultimately promoting apoptotic responses.51 In individuals diagnosed with OSA, the degree of impairment in endothelial-dependent vasodilation correlates with the extent of endothelial cell apoptosis.52 Additionally, reduced apoptosis and increased expression of selectin adhesion molecules have been observed in the polymorphonuclear leukocytes of patients with OSA, suggesting potential effects of the condition on inflammatory and immunological changes in blood leukocytes.53
More accurate pathway detection, achieved through the integration of miRNAs, may facilitate the discovery and development of novel drug targets aligned with pathogenetic mechanisms. Understanding the interplay between the mechanistic pathways and OSA could offer crucial insights for designing adjuvant therapeutic strategies to address the cellular responses to the physiological challenges associated with this medical condition. Nevertheless, our analyses of drug-gene interactions did not reveal any FDA-approved drugs for TFDP2. Furthermore, examination of external datasets suggested that CPAP treatment had no observable impact on the levels of miRNA targets. Therefore, the potential of miRNA-based therapeutics as novel candidates for treatment is particularly relevant considering not only the limited repositioning potential identified in our study but also the promising advances in the use of this technology.13 The regulatory effects of miRNAs on multiple genes of underlying pathways may be advantageous in addressing the multifactorial nature of conditions such as OSA.54 Additional investigations based on current data are therefore warranted.
Strengths and LimitationsThe current investigation has several strengths, including the adoption of a “real clinical context”, multicentric design, comprehensive clinical assessments, the analysis of the plasma miRNome and the employment of diverse feature selection algorithms. However, it is imperative to acknowledge the inherent limitations of this study. Although the discovery phase included a subset of 53 subjects, which may appear limited, this sample size is relatively large compared to typical RNA sequencing studies. Nevertheless, we acknowledge that this limitation may affect the robustness of our findings. To further investigate the relevance of our results, we utilized different external datasets. However, validation in additional cohorts with more diverse demographic and clinical characteristics and an appropriate sample size to ensure adequate statistical power is essential. A significant limitation of our study is the lack of mechanistic investigations to confirm the biological roles of the identified miRNAs and their target genes in the pathogenesis of OSA. While we have established associations between specific miRNAs and OSA using various datasets, experimental studies are vital to elucidate the causal relationships and underlying signaling pathways influenced by these miRNAs. Finally, the criteria for selecting candidate miRNAs in the discovery and technical validation phases were intentionally permissive. This approach was chosen for two reasons. First, the inherent biology of miRNAs allows them to regulate biological responses even with minor fluctuations in their levels,26 which justifies the use of lower fold-change thresholds. Second, the primary goal of these phases was to “feed” the feature selection analysis rather than to identify individual miRNAs specifically associated with OSA.
ConclusionsOur ML feature selection approach identified three plasma miRNAs linked to OSA that, along with their target genes, offer new insights into the underlying pathogenesis of OSA and reveal novel regulatory elements of disease and potential drug targets. These findings hold promise for advancing the development of targeted disease-modifying agents aimed at improving outcomes.
Authors’ ContributionAll authors were involved in the conception and design of the work, the acquisition, analysis and interpretation of the data, as well as in the drafting and critical revision of the work. All authors approved the final version of the manuscript.
Artificial Intelligence InvolvementThe authors declare non artificial intelligence involvement.
Funding StatementThis study was funded by the Instituto de Salud Carlos III (ISCIII) through the projects “PI20/00577” and “PI22/00636” and cofunded by the European Union. DdGC has received financial support from Instituto de Salud Carlos III (Miguel Servet 2020: CP20/00041) cofunded by European Union. FB is supported by the ICREA Academia Program. MCGH held a predoctoral fellowship, “Ayudas al Personal Investigador en Formación”, from IRBLleida/Diputación de Lleida. MM is the recipient of a predoctoral fellowship (PFIS 2021: FI21/00187) from Instituto de Salud Carlos III and cofunded by the European Union. MSdlT has received financial support from the “Ramón y Cajal” grant (RYC2019-027831-I) from the “Ministerio de Ciencia e Innovación—Agencia Estatal de Investigación” cofunded by the European Social Fund (ESF)/“Investing in your future”. CIBERES (CB07/06/2008) is an initiative of the Instituto de Salud Carlos III.
Conflicts of InterestsThe authors declare no competing interests.
Data AvailabilityHTG EdgeSeq miRNA Whole Transcriptome Assay data have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series accession number GSE229362 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE229362).
We thank the individuals who participated in this study, their families, and the clinical and research team in the sleep unit. Human sample manipulation was performed at the Cell Culture Facility, Universitat de Lleida (Lleida, Catalonia, Spain). Work supported by IRBLleida Biobank (B.000682) and Biobank and Biomodels Platform ISCIII PT23/00032.
The followings are the supplementary data to this article:







