


Introducción
El tumor fibroso solitario (TFS) es una neoplasia poco común que representa el 8% de las neoplasias benignas del tórax y el 10% de los tumores pleurales1. El curso clínico de la mayoría de estos tumores es biológicamente benigno, aunque en series de seguimiento prolongado esto no es así2, lo que se ha relacionado con sus características histológicas3. El propósito de este estudio ha sido evaluar las características clinicopatológicas de este tumor en una serie de casos consecutivos procedentes de una misma institución.
Observación clínica
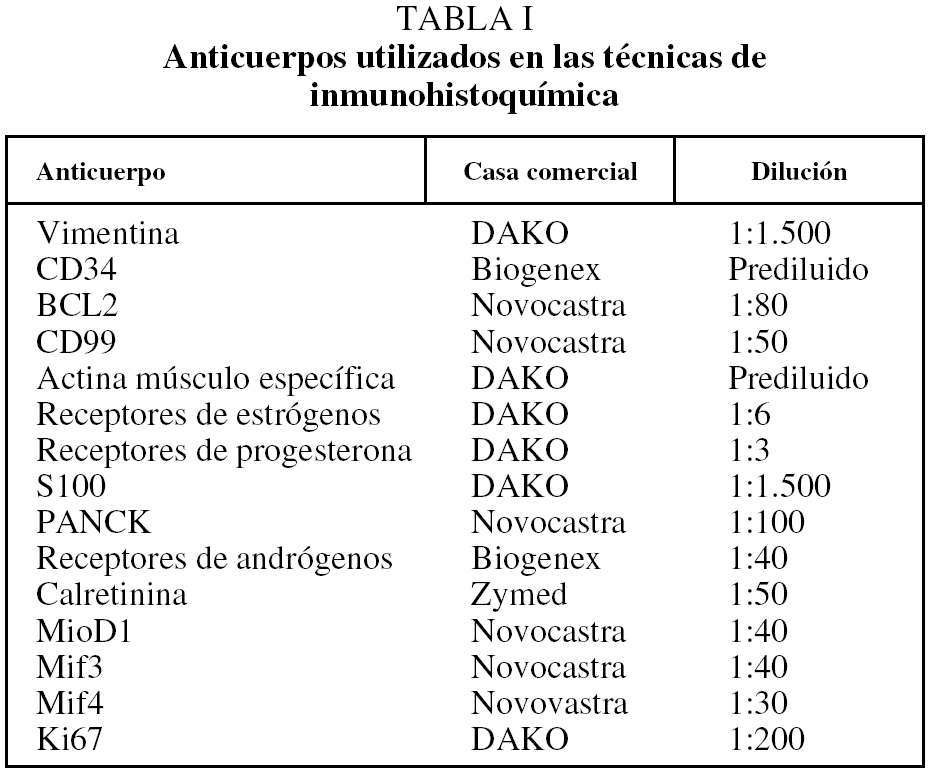
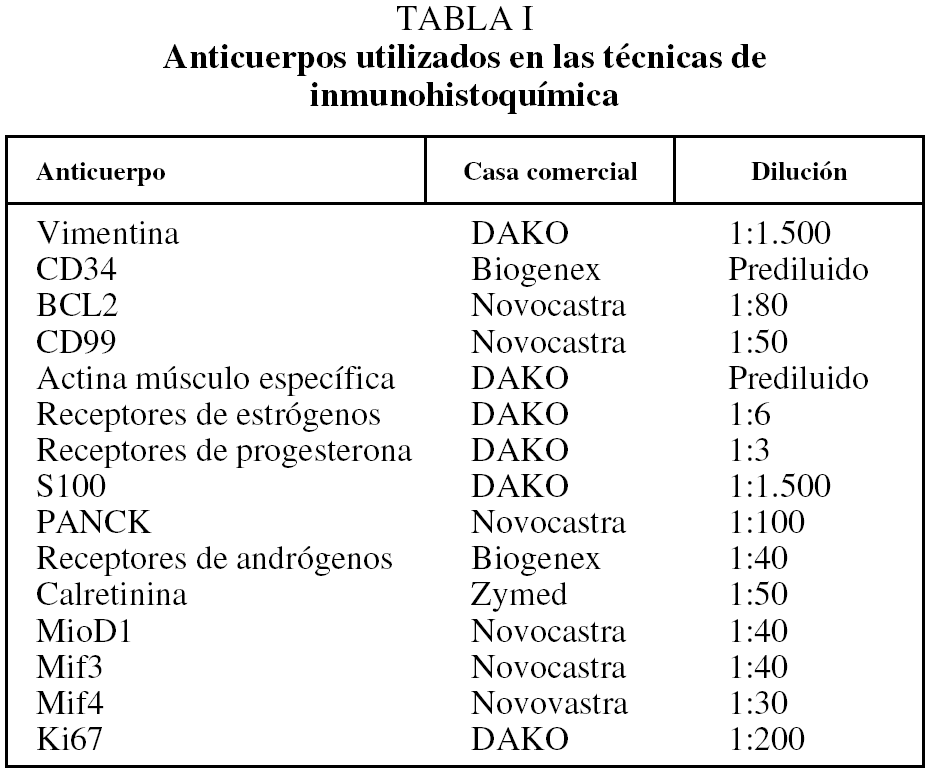
Se ha incluido en el estudio a un total de 30 pacientes con diagnóstico de TFS pleural e intervenidos quirúrgicamente entre 1994 y 2004 en el Hospital General Universitario Gregorio Marañón. Se recogieron las variables sociodemográficas y características clínicas, al igual que el seguimiento. Los casos se clasificaron según los criterios de malignidad establecidos por England et al4. Del total de los casos se obtuvo material para estudio inmunohistoquímico y se realizó seguimiento clínico de los últimos 20 casos. Las tinciones de inmunohistoquímica se realizaron utilizando un panel de anticuerpos monoclonales (tabla I). La inmunorreactividad permitió clasificar en 4 grados según el porcentaje de células teñidas (+++: más del 60% de células; ++: 10-60%; +: menos del 10%; -: ausencia de positividad). El seguimiento de los pacientes se realizó en intervalos de un mes después de la cirugía y posteriormente una vez al año. Los datos se recogieron de las historias médicas.
El promedio de edad fue de 58,39 años (rango: 18-73 años). El 66,6% de los pacientes (20/30) eran mujeres. En el 46,6% de los casos el diagnóstico fue por un hallazgo casual. En los casos que presentaron síntomas la clínica fue de tos en un 10% y de disnea y dolor torácico en un 20%, respectivamente. Ningún caso se asoció a la presencia de síntomas o signos extratorácicos.
La localización en la pleura visceral fue la más frecuente --se observó en un 70% de los casos (21/30)--, seguida de la intrapulmonar, con un 10% (3/30), y la pleura parietal, con un 16,6% (5/30); un caso (3,3%) presentó nódulos tanto en la región de la pleura visceral como intrapulmonar. En cuanto al número de tumoraciones, el 80% de los pacientes presentó tumoración única (fig. 1) y un 20% tumoraciones múltiples; uno de los casos presentó 55 nódulos. El promedio del tamaño tumoral fue de 7,35 cm (rango: 2-21).
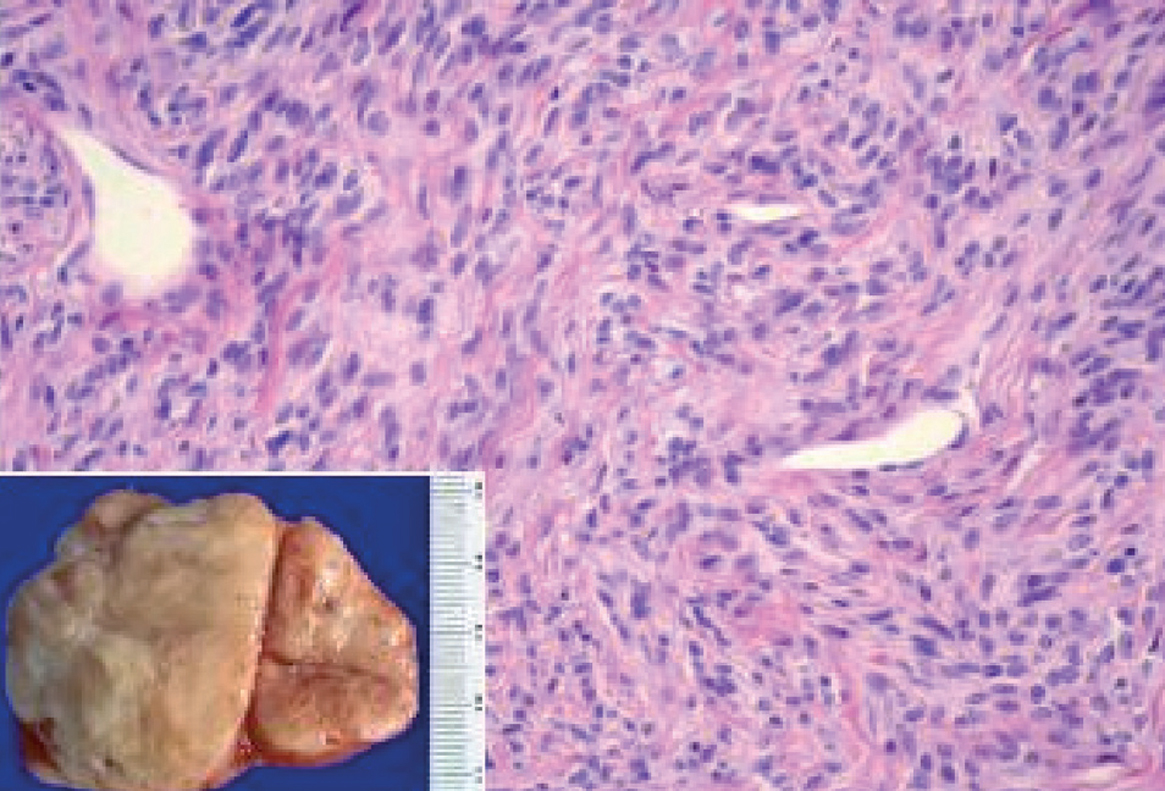
Fig. 1. Macroscopia: tumoración de superficie externa nodular y lisa, de 10 x 9 x 5 cm de ejes mayores y 282 g de peso. La superficie de corte es blanquecina, fasciculada y homogénea. Microscopia: neoplasia fusocelular, que alterna áreas de mayor densidad celular y áreas de escasa densidad celular con abundantes vasos sanguíneos.
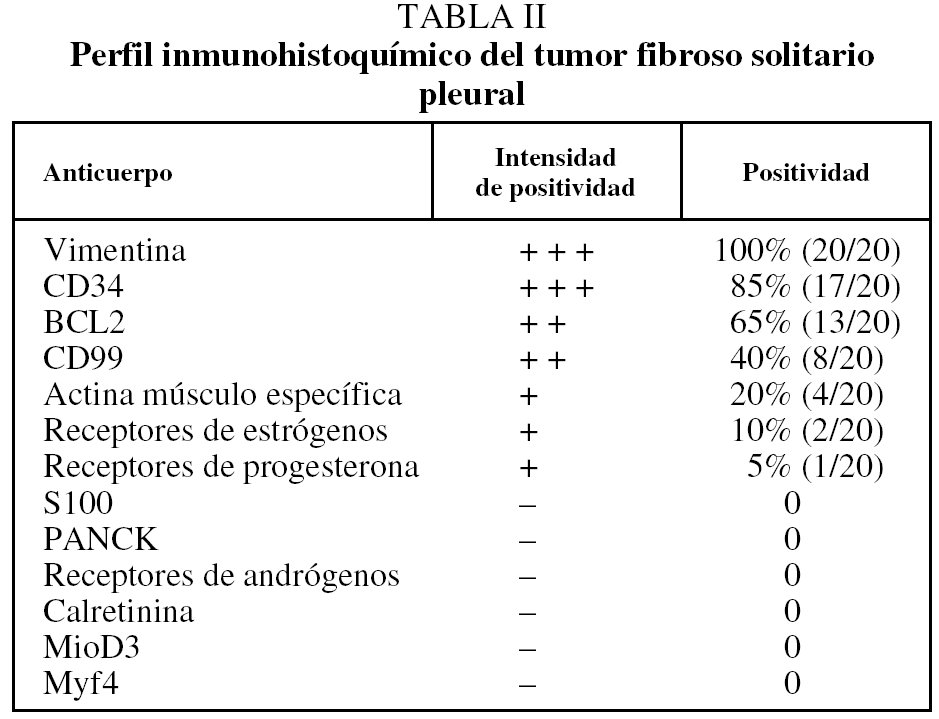
Desde el punto de vista histológico, la densidad celular era escasa en el 15%, moderada en el 50% e intensa en el 35%; no presentaba atipia el 55%, ésta era leve en el 30% y moderada en el 15%; mostraba necrosis el 25%, hemorragia el 15% y mitosis de más de 4 por 10 campos el 30%. En el estudio inmuhistoquímico el 100% presentó positividad para vimentina, el 85% para CD34, el 65% para BCL2 y el 40% para CD99 (40%), y en el 100% las queratinas y la proteína S100 fueron negativas (tabla II y fig. 2).
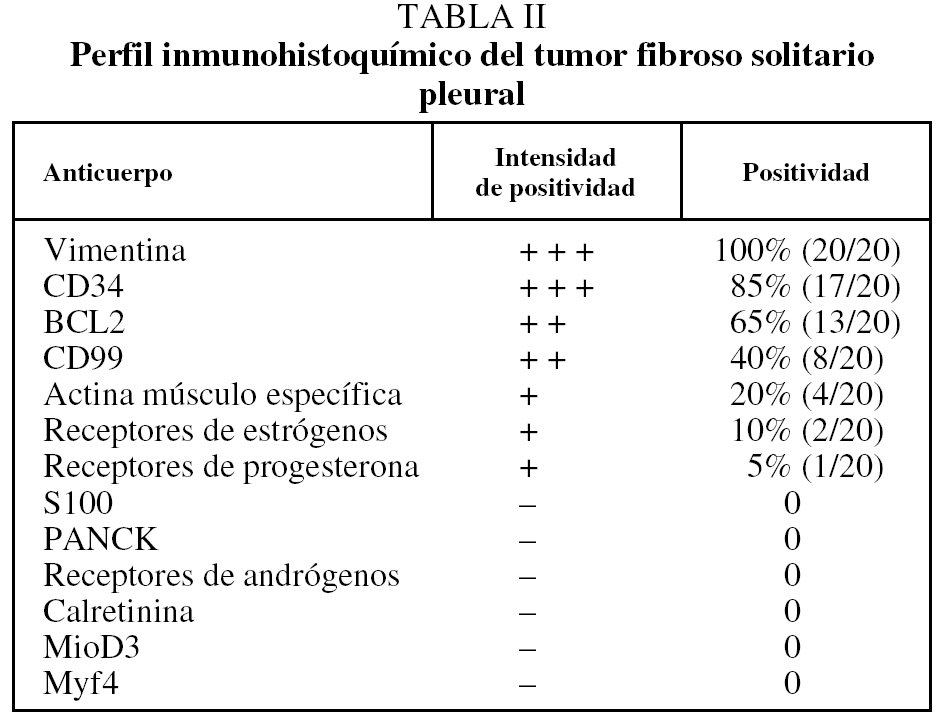
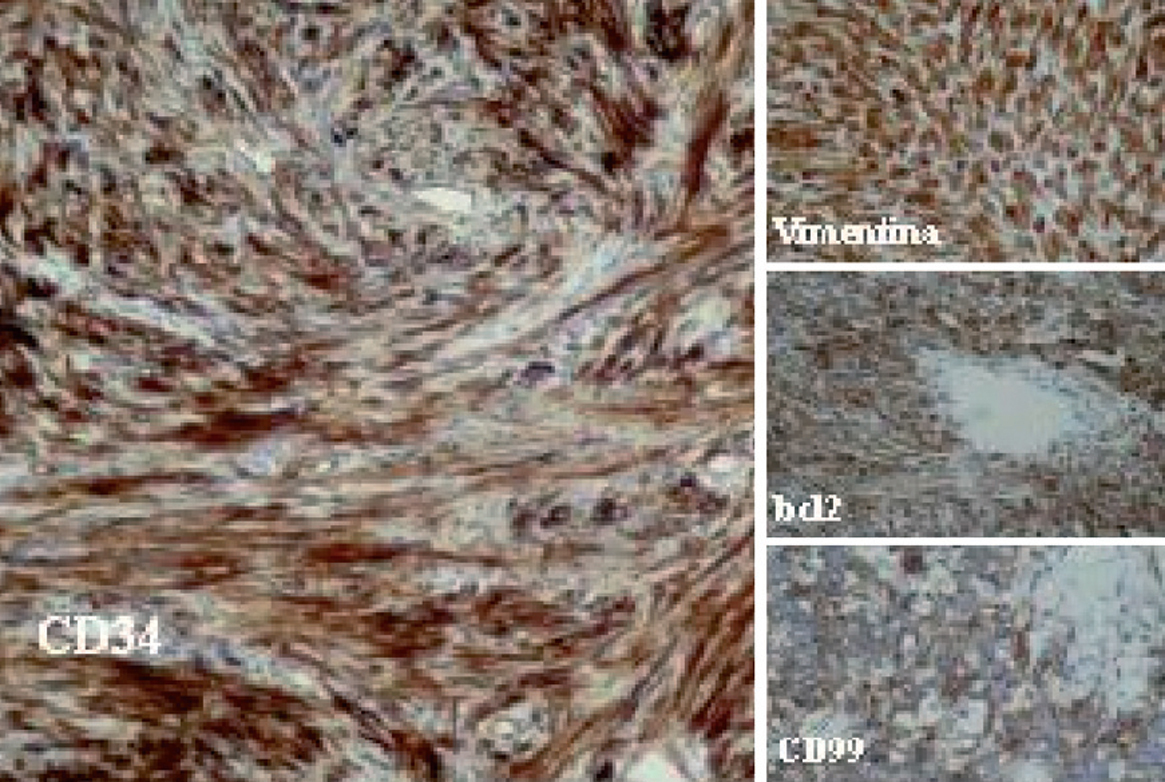
Fig. 2. Técnicas de inmunohistoquímica: positividad para CD34, BCL2, CD99 y vimentina.
El tiempo medio de seguimiento fue 66,32 meses, durante el cual se observó un comportamiento biológico maligno en 4 casos (recurrencia local y metástasis). Estos casos presentaron como signos histológicos indicativos de malignidad los siguientes: densidad celular elevada, atipia de leve a moderada e índice de mitosis superior a 4 por 10 campos, que se correspondió con un Ki67 superior al 10%. De todos los potenciales signos histológicos predictivos de malignidad, sólo el índice de mitosis se asoció a comportamiento maligno (p = 0,008). Es importante mencionar que 2 (50%) de los casos que presentaron comportamiento maligno mostraron CD34 negativo, tanto en el tumor primario como en las recidivas, y uno de ellos tuvo una evolución muy agresiva, con muerte de la paciente. En este caso se realizó el estudio de la translocación SSX/SYT para descartar sarcoma sinovial monofásico, pero fue negativa.
Discusión
El TFS pleural ha recibido una gran variedad de denominaciones. Entre los términos utilizados encontramos los siguientes: mesotelioma localizado, mesotelioma fibroso benigno, fibroma benigno localizado, fibroma submesotelial y fibroma subseroso5. A pesar de que la mayoría de los casos se localizan en la pleura visceral o parietal, hoy día se conoce que aparecen en otras áreas tales como peritoneo6, pericardio6, mediastino7, meninges8, pulmón4, tiroides9, parótida10, órbita11, nariz y fosas nasales12. Esto indica que podría partir de una célula madre común presente en varios órganos y tejidos y que el origen podría ser probablemente miofibroblástico2. En nuestra serie sólo incluimos los casos que se localizaban en la cavidad torácica; de éstos, 4 presentaron lesiones múltiples de localización intrapulmonar, que se asociaron a comportamiento maligno, lo cual podría considerarse un factor de peor pronóstico.
Clínicamente este tumor es asintomático y suele diagnosticarse de manera casual en una radiografía de tórax. Los síntomas locales descritos son tos, disnea, dolor torácico y derrame pleural, y entre las manifestaciones sistémicas figuran artralgias, dedos en "palillo de tambor" e hipoglucemia2-4. En nuestra serie, en el 46,6% de los casos el diagnóstico fue por un hallazgo casual y ninguno se asoció a la presencia de manifestaciones sistémicas.
Según comentan Álvarez y Escalona13, la histogenia de este tumor ha sido un aspecto controvertido que ha estado directamente vinculado con la técnica empleada para su estudio. En 1940 Stout y Murray14, que cultivaron un tumor de aspecto fibrosarcomatoso que crecía en la pleura en cultivos a corto plazo, demostraron que se observaba un doble crecimiento: como células fusiformes y como células poligonales de aspecto epiteliode; éstas se organizaban formando placas, en tanto que las primeras producían un crecimiento radial estrellado a partir de la explantación. Este crecimiento era similar al encontrado por Maximow15 en cultivos de pleura canina, tanto normal como inflamada, y al de los sarcomas sinoviales bifásicos16. No se realizó ningún otro cultivo hasta el trabajo de Sano et al17, quienes observaron en un caso similar que el crecimiento era enteramente superponible. Esto llevó a Stout y Murray a considerar que el tumor fibroso era una variante benigna de las variantes malignas, de modo que también correspondía a un mesotelioma, lo que justificaba el término de mesotelioma fibroso benigno18. Más tarde según explican álvarez y Escalona, se describió el crecimiento diferencial del mesotelioma fibrosarcomatoso, tanto intra como extrapulmonar, característico y propio del mismo, y del sarcoma sinovial, perfectamente diferenciable del crecimiento de aspecto fibroblástico de este tipo de tumores, justificándose con la técnica inicial de descripción la separación de las 2 entidades. Las técnicas inmunohistoquímicas han venido a corroborar dicha concepción, que en un principio se basaba exclusivamente en métodos de microscopia óptica y en cultivos de tejidos.
Un punto importante de discusión es el potencial maligno de este tumor, para lo cual England et al4 establecieron criterios de malignidad, tales como: celularidad aumentada, atipia y pleomorfismo nuclear, índice de mitosis mayor de 4 en 10 campos de gran aumento, áreas grandes de hemorragia y necrosis, derrame pleural, localización atípica e invasión de estructuras adyacentes. De todos los criterios histológicos de malignidad examinados en nuestro estudio, el único que se asoció con peor comportamiento biológico fue el índice de mitosis.
El perfil fenotípico inmunohistoquímico debe establecerse por la combinación de marcadores positivos como vimentina, CD34, BCL2 y CD99 con la negatividad de PANCK y S100.
El diagnóstico diferencial se plantea con el mesotelioma pleural, el sarcoma neurogénico, el sarcoma sinovial, el hemangiopericitoma, el fibrosarcoma y el histiocitoma fibroso maligno. El mesotelioma sarcomatoide muestra una fuerte expresión para las citoqueratinas, que son negativas en el TFS. Los tumores de origen neural son positivos para S100, que en cambio resulta negativo en el TFS. La mayoría de los sarcomas sinoviales monofásicos presentan positividad para citoqueratinas, además de la translocación SSX/SYT típica del sarcoma sinovial19. El estudio de dicha translocación se efectuó en el caso de nuestra serie que presentó evolución rápidamente progresiva, con muerte de la paciente, y resultó negativa. En cuanto al diagnóstico diferencial entre hemangiopericitoma y TFS, hoy día se considera que el primer término debe restringirse sólo para los tumores con diferenciación pericítica (células glómicas/musculares lisas) y que el segundo es una entidad patológica independiente que no se ha relacionado con un origen pericítico20.
Es importante hacer referencia a la ausencia de expresión de CD34 en el TFS, hecho que se observó en 3 casos de nuestra serie; 2 de ellos tuvieron una evolución maligna y uno falleció por progresión tumoral. Por tanto, en el TFS la negatividad del CD34 parece asociarse a un comportamiento biológico más agresivo, dato que se ha señalado en estudios previos y que puede tener utilidad clínica en la predicción de su comportamiento21.
En el 9-19% de los casos se observa recidiva local a pesar de la escisión quirúrgica completa, en el 0-19% aparecen metástasis a distancia y en el 0-27% se produce la muerte en relación con el tumor4,7. En nuestra serie un 20% de los pacientes presentó progresión tumoral a pesar de la escisión quirúrgica y uno de éstos (5%) falleció.
En resumen, el TFS pleural es una neoplasia poco común, usualmente asintomática, cuyo diagnóstico se presenta como un hallazgo casual en una radiografía de tórax. El tratamiento de elección es la escisión quirúrgica completa y, puesto que el comportamiento biológico es impredecible, el seguimiento de los casos debe enfocarse a la detección precoz de la recurrencia local o la aparición de metástasis.
Recibido:23-5-2005; aceptado para su publicación: 27-9-2005.
Correspondencia: Dra. N. Sánchez-Mora.
Hospital General Universitario Gregorio Marañón.
Departamento de Anatomía Patológica.
C/ Dr. Esquerdo, 46. 28007 Madrid. España.
Correo electrónico: histonora@yahoo.es







