
En el presente trabajo se describen las características generales, objetivos y aspectos organizativos de los registros de enfermedades respiratorias minoritarias integrados en el Registro Nacional de Enfermedades Raras del Instituto de Investigación de Enfermedades Raras (IIER), con el objetivo de dar a conocer su existencia y fomentar la participación de los profesionales.
Se recoge información sobre registros de las siguientes enfermedades: déficit de alfa-1 antitripsina, estenosis traqueal idiopática, histiocitosis pulmonar de células de Langerhans del adulto, linfangioleiomiomatosis, proteinosis alveolar y sarcoidosis.
This report describes the general characteristics, objectives and organizational aspects of the registries of rare respiratory diseases included in the National Registry of Rare Diseases of the Research Institute for Rare Diseases (ISCIII), in order to publicize their existence and encourage the participation of professionals.
Information is collected on the following conditions: alpha-1 antitrypsin deficiency, idiopathic tracheal stenosis, adult pulmonary Langerhans’ cell histiocytosis, lymphangioleiomyomatosis, alveolar proteinosis, and sarcoidosis.
La Sociedad Española de Neumología y Cirugía Torácica (SEPAR) tiene una larga tradición y una amplia experiencia en los registros de enfermedades respiratorias, incluyendo algunas minoritarias (ERM)1. Por otro lado, el Instituto de Salud Carlos III (ISCIII) incluyó a las enfermedades raras (ER), como una de sus líneas prioritarias de actuación y fruto de este interés se creó el Instituto de Investigación de Enfermedades Raras (IIER) en 2003 cuyo objetivo es el fomento y ejecución de la investigación clínica y básica, formación y apoyo a la referencia sanitaria e innovación en la atención de la salud en ER.
Las funciones del IIER, entre otras, son las de identificar la magnitud de las ER, estableciendo un sistema de información de base epidemiológica que permita obtener información precisa sobre la prevalencia de estas enfermedades, pero también sobre la distribución de los recursos sanitarios dedicados a ellas. Para ello, se creó en 2005 el Registro Nacional de Enfermedades Raras (RNER) cuya finalidad es el seguimiento, control de la salud e investigación de los pacientes afectados por ER, familiares y población control, que participa en los estudios de investigación. El registro se ha reforzado a través del proyecto SpainRDR de sus siglas en inglés (Spanish Rare Diseases Registry Research Network [Red Española de Registros de Enfermedades Raras para la Investigación]: https://spainrdr.isciii.es) financiado por el ISCIII, al amparo del Consorcio Internacional de investigación de Enfermedades Raras (IRDiRC), constituyendo un proyecto en red dentro de este consorcio internacional (http://www.irdirc.eu y también http://ec.europa.eu/research/health/medical-research/rare-diseases/irdirc_en.html).
Los objetivos comunes de ambas instituciones en las ER motivaron la instauración de un convenio para la promoción de los registros de ERM en 2011. Para ello, se estableció la creación de un grupo de seguimiento o supervisión de los trabajos relacionados con el convenio, la denominación de responsables de cada registro y la designación por parte de SEPAR de la persona responsable de la marcha del convenio. El grupo de responsables de cada ERM incluida en el RNER actúa como coordinador de la red de expertos de dicha patología. Sus funciones son el diseño de las variables específicas y evolutivas, la actividad registral y la promoción de la investigación.
Para el mejor funcionamiento y coordinación de los datos de estas enfermedades en el marco del RNER, los responsables de cada registro y los neumólogos que participan en dichos registros suscriben un reglamento de funcionamiento interno, que sigue las bases y criterios de SEPAR, publicado en febrero de 2013, y las recomendaciones del propio Instituto de Investigación en Enfermedades Raras, relacionadas con las normas de fichero de nivel 3 de seguridad de la Agencia Española de Protección de datos (AEPD). Dicho reglamento también describe las condiciones de explotación de los datos.
Registro Nacional de Enfermedades RarasEl RNER consta de 2 líneas estratégicas principales: los registros de pacientes orientados a resultados clínicos y a la historia natural, y los registros de base poblacional dirigidos a la investigación epidemiológica, socio-sanitaria y a la planificación de temas relacionados con aspectos socio-sanitarios.
Los registros de pacientes pueden estar formalizados a través de convenios con sociedades científicas o a partir de la experiencia de una institución o de una red de expertos1. Uno de los problemas de este tipo de registros, y de la información que suministran, es que los casos incluidos están limitados a los atendidos en los centros participantes, existiendo la posibilidad de incurrir en un sesgo de selección hacia los casos más graves. La notificación de casos es voluntaria, circunscrita a determinados expertos y se requiere el consentimiento del paciente2.
Actualmente las sociedades científicas que han llegado a un acuerdo de colaboración con el ISCIII, y están participando en esta estrategia de desarrollo de registros de pacientes dentro del RNER y en relación también con el SpainRDR, son, además de la SEPAR: la Sociedad Española de Neumología Pediátrica (SENP), la Sociedad Española de Medicina Familiar y Comunitaria (semFYC), la Red Europea de Anemias Congénitas (ENERCA), la Sociedad Española de Alergia e Inmunología Clínica (SEAIC), la Sociedad Española de Endocrinología Pediátrica (SEEP) y la Sociedad Española de Neurología (SEN), así como el Centro de Investigaciones Energéticas, Medioambientales y Tecnológicas (CIEMAT). En la actualidad, se mantienen negociaciones con otras 5 sociedades, además de redes de investigación como el CIBERNED. También se han firmado acuerdos con la industria farmacéutica y la propia Federación Española de Enfermedades Raras (FEDER). A nivel internacional, el consorcio IRDiRC ya ha puesto en marcha proyectos muy relacionados con SpainRDR, como el proyecto RD-CONNECT (http://www.rd-connect.eu) y previamente la DG SANCO de la Comisión Europea había aprobado el proyecto EPIRARE (http://www.epirare.eu). El primero tratará de estandarizar los registros de pacientes, sus biobancos y datos procedentes de investigaciones «omics» a nivel internacional, mientras que el segundo ha definido las bases de la futura plataforma europea de registros de ER que se implantará en los próximos años en el Joint Research Centre (JRC) de la Comisión Europea, localizado en Ispra, Italia. El IIER está implicado en ambos proyectos como socio de los mismos y responsable de actividades como la definición de la calidad, de la selección y definición de las variables comunes a intercambiar y la interoperabilidad entre biobancos.
En cuanto al funcionamiento del RNER y del proyecto SpainRDR, el registro de base poblacional pretende recoger información de todas las ER, a través de la colaboración con todas las comunidades autónomas (CCAA), para lo que se han elaborado unas estrategias de recogida de datos a través de diferentes fuentes de información electrónicas, como el Conjunto Mínimo Básico de Datos (CMBD), que contiene la información procedente de las altas hospitalarias, los registros de mortalidad, las fuentes de atención primaria, los programas de cribado neonatal y malformaciones congénitas, entre otras. En un futuro próximo, esta información, una vez validada y consolidada, permitirá estimar la incidencia de las diferentes enfermedades, su localización más frecuente, la prevalencia y la supervivencia, siempre que se pueda desarrollar un seguimiento adecuado de cada uno de los casos3.
Cada una de las 17 CCAA está elaborando su propio registro autonómico de ER; en el caso de las ciudades autónomas, Ceuta y Melilla, la información se revierte al IIER que se encargará de establecer un registro para ellas. Cada CCAA posteriormente suministrará datos procedentes de las diferentes fuentes de información al RNER.
Además, el RNER ofrece la posibilidad del autorregistro de los pacientes, es decir, cualquier persona afectada por una enfermedad catalogada como rara o sus padres o tutores legales puede registrarse en la web, facilitar información clínica sobre su enfermedad y otorgar su consentimiento informado para que sus datos puedan ser utilizados en investigación. El acceso al RNER se realiza a través de la página: https://registroraras.isciii.es.
Para la creación de los registros de ERM dentro del Registro Nacional, cada comité asesor ha seleccionado las variables relevantes a recoger. Además, se ha estandarizado la recogida de los parámetros comunes (datos demográficos, tabaquismo, pruebas de función respiratoria, gasometría y test de marcha) para poder, en un futuro, comparar datos entre registros. Así mismo, a los parámetros puramente respiratorios se han añadido cuestionarios genéricos de calidad de vida que permitirán comparar el impacto de las ERM con otras ER no respiratorias o incluso con enfermedades comunes.
Todos los registros incluyen información de calidad adecuada a cada proceso, se han adaptado a la legislación vigente sobre protección de datos y serán accesibles tanto desde la web del registro nacional (https://registroraras.isciii.es) como desde la web de la SEPAR (www.separ.es) o desde la propia web del proyecto SpainRDR (https://spainrdr.isciii.es).
Las actualizaciones de los contenidos de cada registro son responsabilidad de cada comité asesor y, en última instancia, de cada coordinador, como representante del comité asesor ante la SEPAR y el IIER.
El RNER no proporciona fondos para el desarrollo de investigaciones paralelas a partir de los datos del registro. Este tipo de desarrollos deberán financiarse, si se precisan fondos para ello, con cargo a ayudas provenientes de la propia sociedad o de agencias de investigación nacionales e internacionales. La incorporación del RNER al concierto internacional de IRDiRC facilitará y promoverá la colaboración con registros similares desarrollados en otros países.
De esta breve descripción se deducen las múltiples aplicaciones de la información recogida y las ventajas que la acumulación de datos de calidad tiene para la investigación biomédica en el momento actual, en el que la experiencia individual de los profesionales sobre una enfermedad se ve reforzada por las opciones tecnológicas de compartir información en aras de mejorar nuestro conocimiento.
Registro Español de Pacientes con Déficit de Alfa-1 antitripsinaEl déficit de alfa-1 antitripsina (AAT) es consecuencia de mutaciones que confieren cambios conformacionales en la proteína AAT que condicionan su función. Se manifiesta principalmente como enfisema pulmonar, cirrosis hepática y, con menor frecuencia, como otras enfermedades (paniculitis, vasculitis…)4. Se estima que en España habría alrededor de 12.000 individuos con déficit grave de AAT5.
El Registro Español de Pacientes con Déficit de Alfa-1 antitripsina (REDAAT), constituido a principios de los años 90, forma parte del área de trabajo de la enfermedad pulmonar obstructiva crónica (EPOC) de la SEPAR. La finalidad del REDAAT desde su fundación es profundizar en el conocimiento sobre el déficit de AAT, contribuir a su difusión, mejorar el tratamiento de las personas afectadas y estimular la investigación6–11.
Los recursos con los que cuenta el REDAAT actualmente son: un comité asesor formado por 12 neumólogos, 3 pediatras y 3 investigadores básicos y cuenta además, con los responsables del laboratorio de referencia y el personal que se encarga del soporte informático. Colaboran en el registro más de 300 médicos de toda España. El principal recurso técnico es www.redaat.es que legalmente pertenece a la Fundación Española de Pulmón-Respira. Incluye la base de datos de los casos de déficit de AAT, cuyo fichero está registrado en la AEPD. Dispone de un área de acceso público con información general y de un área de acceso restringido para profesionales sanitarios, que incluye la ficha de recogida de datos de los pacientes, así como información a tiempo real de los casos registrados y características globales, diagnóstico y tratamiento.
El REDAAT ha colaborado activamente en el registro internacional Alpha One International Registry ([AIR] http://www.antitrypsindeficiency.org) desde su formación, y con algunos registros nacionales europeos12,13.
Dado que este registro dispone de su propio dominio y una base de datos en funcionamiento, creada con anterioridad a la constitución del RNER, no está integrado en la plataforma de este registro sino que comparte la información con él.
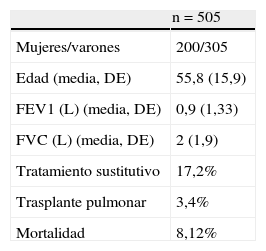
En la tabla 1 se resumen las características de los casos registrados.
Datos basales de la población de pacientes incluida en el Registro Español de Pacientes con Déficit de Alfa-1 antitripsina (2001-2013)
| n=505 | |
| Mujeres/varones | 200/305 |
| Edad (media, DE) | 55,8 (15,9) |
| FEV1 (L) (media, DE) | 0,9 (1,33) |
| FVC (L) (media, DE) | 2 (1,9) |
| Tratamiento sustitutivo | 17,2% |
| Trasplante pulmonar | 3,4% |
| Mortalidad | 8,12% |
DE: desviación estándar; FEV1: volumen espiratorio máximo en el primer segundo; FVC: capacidad vital forzada; L: litros.
Dentro de la denominada estenosis traqueal se pueden definir 2 grupos: la estenosis laringotraqueal benigna secundaria y la idiopática14.
La estenosis traqueal idiopática es una enfermedad caracterizada por el desarrollo de una estenosis cicatricial fibrosa a nivel del cricoides y el tercio superior de la tráquea, con presencia de engrosamiento de la submucosa, sin afectación del cartílago subyacente. Afecta mayoritariamente a mujeres y los síntomas principales son disnea progresiva y estridor que se desarrollan a lo largo de años. Su diagnóstico es incompatible con la presencia de otros antecedentes relacionados con estenosis de la vía aérea superior que definirían la estenosis secundaria15.
Su incidencia y prevalencia son desconocidas, al igual que la etiología y los factores pronósticos. Tampoco se dispone de normativas específicas sobre su diagnóstico y tratamiento, ni todavía se ha incluido en los catálogos internacionales de ER. La experiencia acumulada en esta enfermedad se centra, principalmente, en las diferentes técnicas quirúrgicas empleadas para su tratamiento16–21.
Las estenosis laringotraqueales benignas secundarias suelen estar motivadas por una de las siguientes causas: postintubación, postraumática, por inhalación de humos tóxicos, posquirúrgica, infecciosa, inflamatoria, compresión extrínseca y neurológica, y no son el objetivo de este registro14.
En los últimos 30 años la cirugía traqueal, junto con otras alternativas terapéuticas (láser, dilataciones y prótesis traqueales), ha conseguido que la mayoría de estas lesiones benignas laringotraqueales puedan ser tratadas, si bien su relativa rareza limita la experiencia acumulada por los centros asistenciales.
El Registro Español de Estenosis Traqueal (REET) está integrado en el Área de Cirugía Torácica de la SEPAR. Más de 40 profesionales se han inscrito en el grupo de colaboradores de este registro. Su base de datos está integrada en la plataforma del RNER y la recogida de casos se iniciará en 2014.
Registro Español de Histiocitosis Pulmonar de Células de LangerhansLa histiocitosis pulmonar de células de Langerhans (HPCL) en el adulto se caracteriza por una proliferación peribronquiolar del histiocito de Langerhans, célula dendrítica que participa en la respuesta inmune. Sin preferencia de género, afecta fundamentalmente a adultos entre 20 y 40 años de edad. La evolución es en general benigna, aunque cerca de un 30% presenta un progresivo deterioro de la función pulmonar. Si bien se relaciona con el hábito tabáquico, puesto que el 95% de los afectados son fumadores, la etiología no está aclarada, como tampoco la incidencia ni la prevalencia exacta de la HPCL en adultos.
Recientemente, se ha publicado un documento de consenso de expertos que recoge las recomendaciones sobre el diagnóstico y tratamiento de esta enfermedad, que se espera que facilite la estandarización de los procedimientos22. El principal tratamiento, en casos graves, es el trasplante pulmonar aunque se ha propuesto el uso de quimioterápicos para tratar la progresión de la enfermedad.
En nuestro entorno existen pocos datos sobre las características de la HPCL y su manejo. Recientemente se ha publicado la primera serie multicéntrica, que demuestra la importancia del uso de guías clínicas para el diagnóstico y tratamiento de la enfermedad, como la anteriormente propuesta23. Además, profundiza en el interrogante del tabaco como desencadenante de la enfermedad y la necesidad de indagar en otras posibles vías fisiopatológicas.
El Registro Español de Histiocitosis Pulmonar de Células de Langerhans (REHPCL) forma parte del programa integrado de investigación de enfermedades intersticiales difusas de la SEPAR (PII-EPID). Consta de un comité asesor formado por 5 expertos, su base de datos está ubicada en la plataforma del RNER y se ha iniciado la recogida de casos en septiembre de 2013. Este registro, además de una vertiente puramente epidemiológica, tiene como objetivo desarrollar paralelamente proyectos de investigación que favorezcan la investigación translacional sobre esta patología. En este contexto, nació el proyecto Caracterización funcional y genética de la HPCL, en el que participan diversos investigadores de la SEPAR y del IIER (ISCIII). El proyecto ha recibido una beca de la Fundación Mutua Madrileña y del PII-EPID (Ayudas a la investigación EPID-Futuro).
Registro Español de LinfangioleiomiomatosisLa linfangioleiomiomatosis (LAM) es una enfermedad pulmonar que afecta a mujeres jóvenes en edad fértil. Se caracteriza por un crecimiento anómalo de células musculares lisas atípicas (células LAM) a nivel pulmonar, incluyendo las vías aéreas y los vasos linfáticos y sanguíneos, que da lugar a la formación de quistes, lo que finalmente conlleva una destrucción del parénquima pulmonar. Se distinguen 2 tipos de LAM: una forma esporádica con afectación exclusivamente pulmonar (LAM esporádica) y otra asociada al complejo esclerosis tuberosa, que es un síndrome neurocutáneo autosómico dominante, ligado a mutaciones en los genes TSC1 y TSC2.
Debido a su baja prevalencia es una enfermedad bastante desconocida. Muchos de los síntomas de la enfermedad (disnea, tos) son similares a los de otras enfermedades pulmonares como el asma o la bronquitis crónica, lo que hace que se diagnostique tardíamente.
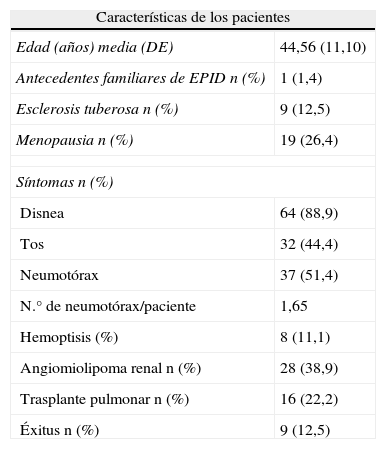
No se conoce la verdadera incidencia de la LAM. En Estados Unidos, la sociedad americana de LAM (The LAM foundation) tiene 1.300 pacientes registradas. En España, el grupo de trabajo de Enfermedades Pulmonares Intersticiales Difusas (EPID) de la SEPAR en colaboración con la Asociación Española de Pacientes con LAM (AELAM), recogió 72 casos procedentes de 23 centros de 8 CCAA y Andorra, en un estudio transversal, como base del Registro Español de LAM (RELAM). Las características generales recogidas en este registro se describen en la tabla 224.
Características de las pacientes con LAM
| Características de los pacientes | |
| Edad (años) media (DE) | 44,56 (11,10) |
| Antecedentes familiares de EPID n (%) | 1 (1,4) |
| Esclerosis tuberosa n (%) | 9 (12,5) |
| Menopausia n (%) | 19 (26,4) |
| Síntomas n (%) | |
| Disnea | 64 (88,9) |
| Tos | 32 (44,4) |
| Neumotórax | 37 (51,4) |
| N.° de neumotórax/paciente | 1,65 |
| Hemoptisis (%) | 8 (11,1) |
| Angiomiolipoma renal n (%) | 28 (38,9) |
| Trasplante pulmonar n (%) | 16 (22,2) |
| Éxitus n (%) | 9 (12,5) |
DE: desviación estándar; EPID: enfermedad pulmonar intersticial difusa; LAM: linfangioleiomiomatosis; NS/NC: no sabe/no contesta.
Fuente: Modificada de Antón et al.23.
El RELAM está integrado dentro del PII-EPID y consta de un comité asesor compuesto por 8 personas. Su base de datos está ubicada en la plataforma del RNER y la recogida de casos se ha iniciado en septiembre del 2013.
Registro Español de Proteinosis AlveolarLa proteinosis alveolar pulmonar (PAP) es una enfermedad caracterizada por una acumulación alveolar de proteínas y lípidos del surfactante, que produce una alteración en el intercambio gaseoso y puede desencadenar insuficiencia respiratoria.
Existen 3 formas de PAP: la autoinmune o adquirida, secundaria y congénita.
La primera representa el 90% de las PAP del adulto, comienza alrededor de la 5.a década, es más frecuente en hombres (2:1) y presenta anticuerpos antiestimuladores de colonias granulocíticas (anti-GM-CSF). La forma secundaria, que completa el 10% restante de las PAP del adulto, con una edad de inicio similar a la autoinmune, es más frecuente en hombres (1,5:1) que padecen enfermedades hematológicas (especialmente síndrome mielodisplásico), infecciones respiratorias o enfermedades autoinmunes, cursa con anti-GM-CSF negativos y tiene un peor pronóstico que las de origen autoinmune (supervivencia media a 2 años del 50% y a 5 años del 35%). Finalmente se conoce la congénita que suele asociarse a mutaciones de los genes que codifican las proteínas del surfactante pulmonar.
La PAP en general tiene una prevalencia de 3,7 pacientes por millón de habitantes, si bien depende de la zona geográfica al tener una asociación racial.
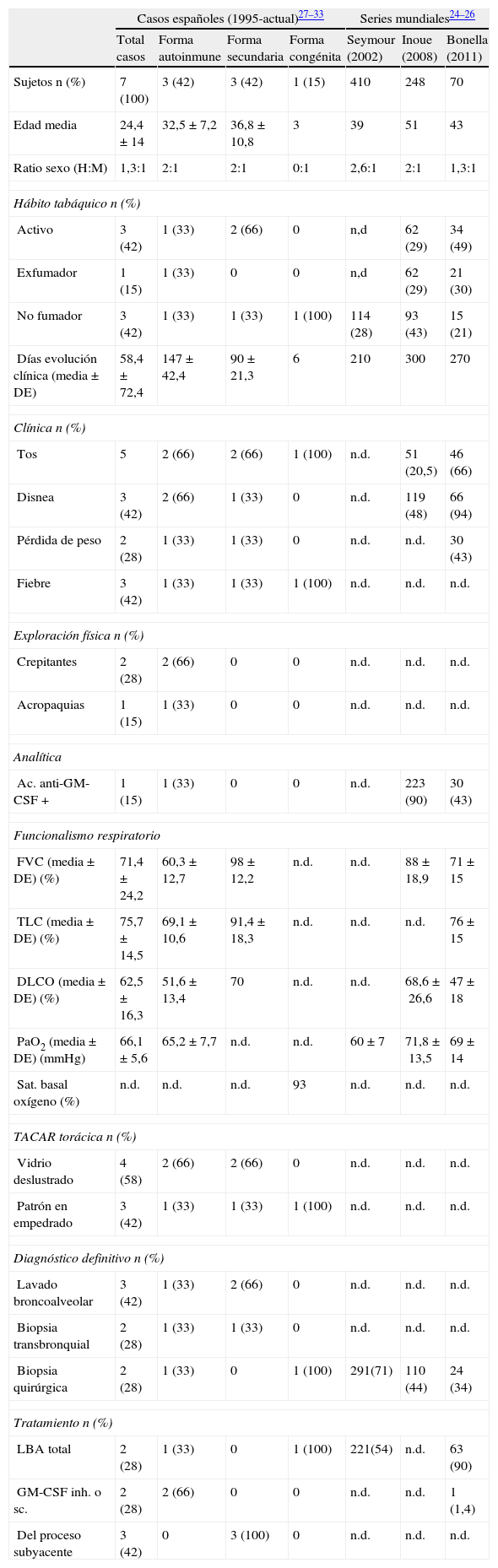
Se desconoce el número total de casos existentes en España. Los resultados más significativos de las series publicadas tanto a nivel nacional como internacional se resumen en la tabla 325–34.
Datos clínicos de los casos españoles de PAP publicados desde 1995 hasta la fecha, diferenciándolos según etiología. Resumen de las grandes series mundiales de PAP publicadas hasta la fecha
| Casos españoles (1995-actual)27–33 | Series mundiales24–26 | ||||||
| Total casos | Forma autoinmune | Forma secundaria | Forma congénita | Seymour (2002) | Inoue (2008) | Bonella (2011) | |
| Sujetos n (%) | 7 (100) | 3 (42) | 3 (42) | 1 (15) | 410 | 248 | 70 |
| Edad media | 24,4±14 | 32,5±7,2 | 36,8±10,8 | 3 | 39 | 51 | 43 |
| Ratio sexo (H:M) | 1,3:1 | 2:1 | 2:1 | 0:1 | 2,6:1 | 2:1 | 1,3:1 |
| Hábito tabáquico n (%) | |||||||
| Activo | 3 (42) | 1 (33) | 2 (66) | 0 | n,d | 62 (29) | 34 (49) |
| Exfumador | 1 (15) | 1 (33) | 0 | 0 | n,d | 62 (29) | 21 (30) |
| No fumador | 3 (42) | 1 (33) | 1 (33) | 1 (100) | 114 (28) | 93 (43) | 15 (21) |
| Días evolución clínica (media±DE) | 58,4±72,4 | 147±42,4 | 90±21,3 | 6 | 210 | 300 | 270 |
| Clínica n (%) | |||||||
| Tos | 5 | 2 (66) | 2 (66) | 1 (100) | n.d. | 51 (20,5) | 46 (66) |
| Disnea | 3 (42) | 2 (66) | 1 (33) | 0 | n.d. | 119 (48) | 66 (94) |
| Pérdida de peso | 2 (28) | 1 (33) | 1 (33) | 0 | n.d. | n.d. | 30 (43) |
| Fiebre | 3 (42) | 1 (33) | 1 (33) | 1 (100) | n.d. | n.d. | n.d. |
| Exploración física n (%) | |||||||
| Crepitantes | 2 (28) | 2 (66) | 0 | 0 | n.d. | n.d. | n.d. |
| Acropaquias | 1 (15) | 1 (33) | 0 | 0 | n.d. | n.d. | n.d. |
| Analítica | |||||||
| Ac. anti-GM-CSF + | 1 (15) | 1 (33) | 0 | 0 | n.d. | 223 (90) | 30 (43) |
| Funcionalismo respiratorio | |||||||
| FVC (media±DE) (%) | 71,4±24,2 | 60,3±12,7 | 98±12,2 | n.d. | n.d. | 88±18,9 | 71±15 |
| TLC (media±DE) (%) | 75,7±14,5 | 69,1±10,6 | 91,4±18,3 | n.d. | n.d. | n.d. | 76±15 |
| DLCO (media±DE) (%) | 62,5±16,3 | 51,6±13,4 | 70 | n.d. | n.d. | 68,6±26,6 | 47±18 |
| PaO2 (media±DE) (mmHg) | 66,1±5,6 | 65,2±7,7 | n.d. | n.d. | 60±7 | 71,8±13,5 | 69±14 |
| Sat. basal oxígeno (%) | n.d. | n.d. | n.d. | 93 | n.d. | n.d. | n.d. |
| TACAR torácica n (%) | |||||||
| Vidrio deslustrado | 4 (58) | 2 (66) | 2 (66) | 0 | n.d. | n.d. | n.d. |
| Patrón en empedrado | 3 (42) | 1 (33) | 1 (33) | 1 (100) | n.d. | n.d. | n.d. |
| Diagnóstico definitivo n (%) | |||||||
| Lavado broncoalveolar | 3 (42) | 1 (33) | 2 (66) | 0 | n.d. | n.d. | n.d. |
| Biopsia transbronquial | 2 (28) | 1 (33) | 1 (33) | 0 | n.d. | n.d. | n.d. |
| Biopsia quirúrgica | 2 (28) | 1 (33) | 0 | 1 (100) | 291(71) | 110 (44) | 24 (34) |
| Tratamiento n (%) | |||||||
| LBA total | 2 (28) | 1 (33) | 0 | 1 (100) | 221(54) | n.d. | 63 (90) |
| GM-CSF inh. o sc. | 2 (28) | 2 (66) | 0 | 0 | n.d. | n.d. | 1 (1,4) |
| Del proceso subyacente | 3 (42) | 0 | 3 (100) | 0 | n.d. | n.d. | n.d. |
DE: desviación estándar; DLCO: test de transferencia de monóxido de carbono; FVC: capacidad vital forzada; GM-CSF: factor estimulante de crecimiento de colonias de granulocitos-monocitos; inh: inhalado; LBA: lavado broncoalveolar; n.d.: dato no disponible; PaO2: presión arterial de oxígeno; PAP: proteinosis alveolar pulmonar; Sat: saturación; sc: subcutáneo; TACAR: tomografía axial computerizada de alta resolución; TLC: capacidad pulmonar total.
El Registro Español de Proteinosis Alveolar (REPA) está integrado en el PII-EPID de la SEPAR y su comité asesor está formado por 5 neumólogos. La base de datos se encuentra en la plataforma del RNER y el registro de casos se ha iniciado en septiembre de 2013.
Registro Español de SarcoidosisLa sarcoidosis en una enfermedad granulomatosa sistémica que afecta principalmente a pulmón (90%) y tejido linfático, pudiendo afectar a otros órganos35.
La incidencia y prevalencia global varían en función de la región geográfica analizada, hallándose más de 50/100.000 casos en países nórdicos, mientras que en otros como Japón o España la prevalencia descrita es de<10/100.000 habitantes, con una alta frecuencia del síndrome de Löfgren (48%)36. Afecta principalmente a adultos de edad media. Las grandes series de pacientes, estudiadas durante los últimos 30 años, indican que es probable que sea necesaria una combinación de factores ambientales predisponentes y una genética susceptible para desarrollar la enfermedad, aunque no hay estudios concluyentes37,38. Diferentes publicaciones en la literatura señalan la relevancia de algunos agentes infecciosos (bacterias, virus, hongos) así como la exposición a ambientes rurales.
Uno de los principales problemas clínicos es saber si existe actividad inflamatoria, siendo la dosificación de la enzima conversora de la angiotensina (ECA) la más estudiada, si bien la hipoergia de las pruebas cutáneas retardadas y la linfopenia en sangre periférica también se han utilizado39. Por otra parte se ha observado que los pacientes que padecen esta enfermedad presentan disfunciones inmunológicas en el reconocimiento, procesamiento y presentación de los potenciales antígenos, por los macrófagos y linfocitos T40.
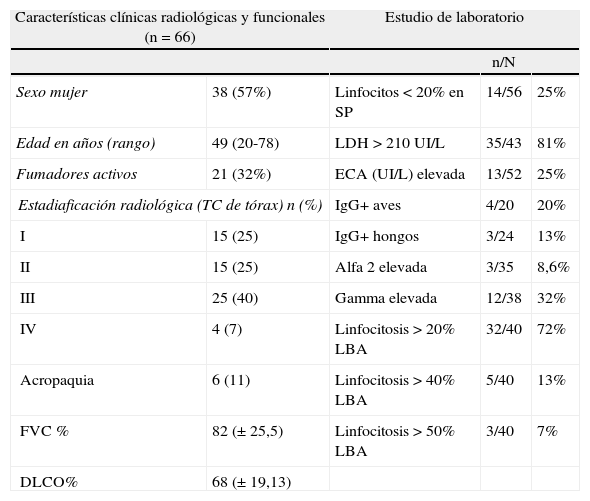
En la tabla 4 se describen los datos generales de la mayor serie española publicada y en la tabla 5 se presentan las características de la serie del Hospital Vall d’Hebron (años 2005-2012) que constituye el punto de partida del registro41.
Casuística de la sarcoidosis del Hospital Universitario Vall d’Hebron (2004-2012)
| Características clínicas radiológicas y funcionales (n=66) | Estudio de laboratorio | |||
| n/N | ||||
| Sexo mujer | 38 (57%) | Linfocitos<20% en SP | 14/56 | 25% |
| Edad en años (rango) | 49 (20-78) | LDH >210 UI/L | 35/43 | 81% |
| Fumadores activos | 21 (32%) | ECA (UI/L) elevada | 13/52 | 25% |
| Estadiaficación radiológica (TC de tórax) n (%) | IgG+ aves | 4/20 | 20% | |
| I | 15 (25) | IgG+ hongos | 3/24 | 13% |
| II | 15 (25) | Alfa 2 elevada | 3/35 | 8,6% |
| III | 25 (40) | Gamma elevada | 12/38 | 32% |
| IV | 4 (7) | Linfocitosis >20% LBA | 32/40 | 72% |
| Acropaquia | 6 (11) | Linfocitosis >40% LBA | 5/40 | 13% |
| FVC % | 82 (± 25,5) | Linfocitosis >50% LBA | 3/40 | 7% |
| DLCO% | 68 (± 19,13) | |||
Alfa 2 elevada: >10,7%; DLCO: test de transferencia de monóxido de carbono; ECA: enzima conversora de la angiotensina; FVC: capacidad vital forzada; Gammaglobulina elevada: > 19,3%; IgG: inmunoglobulina G; LBA: lavado broncoalveolar; LDL: lipoproteína de baja densidad; SP: sangre periférica; TC: tomografía computerizada.
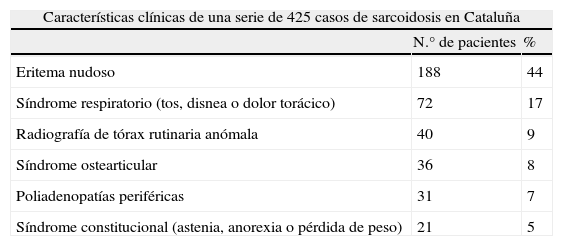
Sarcoidosis en Cataluña: análisis de 425 casos
| Características clínicas de una serie de 425 casos de sarcoidosis en Cataluña | ||
| N.° de pacientes | % | |
| Eritema nudoso | 188 | 44 |
| Síndrome respiratorio (tos, disnea o dolor torácico) | 72 | 17 |
| Radiografía de tórax rutinaria anómala | 40 | 9 |
| Síndrome ostearticular | 36 | 8 |
| Poliadenopatías periféricas | 31 | 7 |
| Síndrome constitucional (astenia, anorexia o pérdida de peso) | 21 | 5 |
Fuente: Modificada de Badrinas et al.41.
El Registro Español de Sarcoidosis (RESAR) está integrado dentro del PII-EPID y su comité asesor está formado por 7 médicos. La base de datos se encuentra en la plataforma del RNER y el registro de casos se ha iniciado en septiembre de 2013.
Aunque el desarrollo inicial de todos los registros haya sido llevado a cabo por un grupo reducido de expertos en cada enfermedad, son una herramienta colaborativa en la que cualquier clínico podrá notificar su caso y también cualquier investigador podrá beneficiarse de la explotación de la información contenida en los mismos, previa solicitud al comité científico.
DiscusiónEn el presente artículo, se proporciona información de los registros de ERM desarrollados en España, en coordinación con el RNER.
Su valor radica en la acumulación de información de los escasos casos existentes y permitirá a la comunidad científica profundizar en el conocimiento de estas enfermedades, pero también tendrá utilidad para los clínicos no expertos en ellas ya que dispondrán de información actualizada y acceso a los expertos en el caso de encontrarse con un paciente afectado. Así mismo, tendrá utilidad para las instituciones sanitarias en la adecuación de los recursos para la asistencia a los pacientes al disponer de información actualizada y dinámica de todo el territorio.
La incorporación de la información recogida por los registros de base poblacional de cada CCAA permitirá tener una visión más próxima a la realidad de la práctica clínica diaria que la que pueden aportar registros de participación voluntaria.
Al igual que en los ensayos clínicos, la calidad de los datos recogidos es fundamental para determinar la validez de los resultados extraídos. En los registros, como estudios observacionales, existe el riesgo de que se produzcan sesgos de muchos tipos, aunque los más frecuentes son los que afectan a la selección de casos y a la calidad de la información disponible, sobre todo de las variables principales (precisión del diagnóstico y su codificación)42. En el caso del RNER, la posibilidad de poder contrastar los datos procedentes de los registros de pacientes con los aportados por los de las CCAA permitirá conocer la representatividad de la muestra y la validez de los resultados a partir de ella.
El papel de los registros en la toma de decisiones sobre la eficacia de los tratamientos es todavía controvertido. No obstante, su papel en la fase de postautorización de los tratamientos, para el seguimiento de efectos secundarios cuando estos son superiores al ensayo clínico y se recogen en un mayor número de casos, así como el análisis de coste-efectividad de dichos tratamientos es fundamental y no puede ser suplantado por un diseño tradicional tipo ensayo clínico43–45.
El RNER del IIER (ISCIII) está incluido en la Estrategia de Enfermedades Raras del Sistema Nacional de Salud. Es una pieza clave de la misma al servir como fuente de información para el desarrollo de políticas socio-sanitarias de planificación y para la investigación. La contribución de todas las CCAA, junto a las sociedades de profesionales, la industria y los propios pacientes, hace de esta herramienta una oportunidad única de cohesión del sistema y proporciona enormes posibilidades de cooperación internacional.
Autoría/colaboradores
Beatriz Lara: redacción del artículo y administradora del REDAAT; Manuel Posada e Ignacio Abaitua: redacción del artículo y responsables del diseño y puesta en funcionamiento del RNER; Genaro Galán: redacción del artículo y coordinador del REET; Diego Castillo: redacción del artículo y coordinador del REHPCL; Álvaro Casanova: redacción del artículo y coordinador del RELAM; Esteban Cano: redacción del artículo y coordinador del REPA; Íñigo Ojanguren: redacción del artículo y coordinador del RESAR.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses y han participado por igual en la redacción de este trabajo.
Los autores quieren agradecer su extraordinaria labor a la Asociación Española de Pacientes con Déficit de Alfa-1 antitripsina, representada por la Sra. Shane Fitch y el Sr. Mariano Pastor y a la Asociación Española de Pacientes con Linfangioleoimiomatosis, representada por la Sra. Asunción Valdivielso y la Sra. M.a Luz Vila.
También agradecen al Dr. Ruiz Manzano, presidente de la SEPAR en 2011, su apoyo a este proyecto desde sus inicios, sin el cual no habría sido posible que viera la luz.







