









Growing evidence shows a hypercoagulable state in obstructive sleep apnea (OSA) that could be a risk factor for thromboembolic disease.
ObjectivesWe aimed to elucidate mechanisms involved in the procoagulant profile observed in patients with OSA and to investigate the potential utility of global tests in its characterization.
MethodsThirty-eight patients with severe OSA without previous history of thrombosis and nineteen healthy age- and sex-matched controls were included.
Kinetic of clot formation was determined using rotational thromboelastometry.
Haemostatic capacity of plasma and microparticles was determined by Calibrated Automated Thrombinography.
Platelet surface receptors, activation markers and formation of platelet/leukocytes aggregates were analyzed by flow cytometry.
ResultsThromboelastometry showed a procoagulant state in patients with OSA that did not seem to be related to a basal activation of platelets but by the increased existence of platelet/leukocyte aggregates.
Patients with OSA presented many signs of endothelial damage such as increased plasma levels of E-selectin and cfDNA and enhanced thrombin generation due to the presence of microparticles rich in tissue-factor, which is related to OSA severity.
ConclusionsOSA induces an enhancement in the dynamics of clot formation which appears to be caused by at least two pathological mechanisms. First, a greater formation of platelet-leukocyte aggregates; secondly, endothelial damage which provokes a greater procoagulant potential due to the increase in tissue factor-rich microparticles. Moreover, this study has identified thromboelastometry and thrombin generation assay as useful tools to evaluate the prothrombotic state in these patients.
Existen pruebas crecientes que muestran un estado de hipercoagulabilidad en la apnea obstructiva del sueño (AOS) que podría ser un factor de riesgo de desarrollar enfermedad tromboembólica.
ObjetivosNuestro objetivo fue dilucidar los mecanismos involucrados en el perfil procoagulante que se ha observado en los pacientes con AOS e investigar la posible utilidad de las pruebas globales en su caracterización.
MétodosSe incluyeron 38 pacientes con AOS grave sin antecedentes de trombosis y 19 controles sanos emparejados por edad y sexo.
La cinética de la formación del coágulo se determinó mediante tromboelastometría rotacional.
La capacidad hemostática del plasma y las micropartículas se determinó mediante trombinografía automatizada calibrada.
Los receptores de la membrana plaquetaria, los marcadores de activación plaquetaria y la formación de agregados de plaquetas-leucocitos se analizaron mediante citometría de flujo.
ResultadosLa tromboelastometría mostró un estado procoagulante en pacientes con AOS que no parecía estar relacionado con una activación basal de las plaquetas, sino por el aumento de agregados de plaquetas-leucocitos.
Los pacientes con AOS presentaban muchos signos de daño endotelial, como un aumento de los niveles plasmáticos de E-selectina y ADNcf y una mayor generación de trombina debido a la presencia de micropartículas ricas en factor tisular, que se relaciona con la gravedad de la AOS.
ConclusionesLa AOS induce un aumento de la dinámica de la formación de coágulos que parece estar causada por al menos 2 mecanismos patológicos. Primero, una mayor formación de agregados plaquetas-leucocitos; segundo, el daño endotelial que provoca un mayor potencial procoagulante debido al aumento de micropartículas ricas en factor tisular. Además, este estudio ha identificado la tromboelastometría y el ensayo de generación de trombina como herramientas útiles para evaluar el estado protrombótico en estos pacientes.
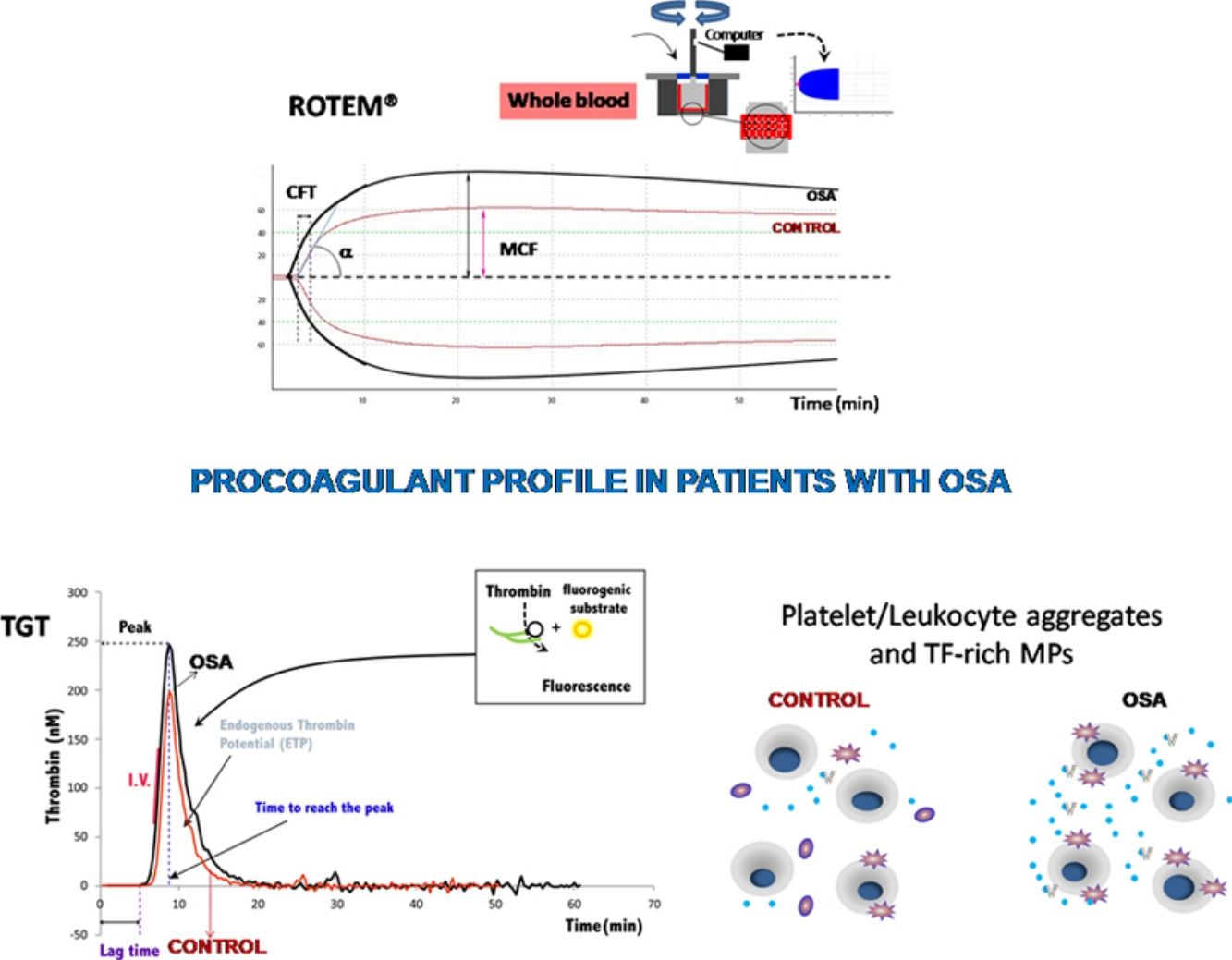
Obstructive sleep apnea (OSA) is a highly prevalent disorder characterized by repetitive episodes of partial or complete upper airway closure during sleep leading to intermittent hypoxia and sleep fragmentation.1 These immediate alterations trigger a cascade of mechanisms, such as increased sympathetic activity, altered vascular regulation, endothelial dysfunction, oxidative stress, and chronic systemic inflammation, which increase the risk of cardiovascular disturbances.2 In fact, OSA is associated with a high incidence of arterial hypertension, stroke, ischemic heart disease, arrhythmias, and heart failure.3
There is growing evidence that OSA could also be a risk factor for venous thromboembolic disease (VTD).4 This association represents a major public health burden due to its high prevalence and mortality. The overall annual incidence rate of VTD reaches 75–269 cases per 100,000 persons, and its risk approximately doubles with each decade after the age of 40 years.5 Moreover, pulmonary embolism, the primary manifestation of VTD, is the third most frequent cardiovascular disease.5
Although some risk factors for OSA, including obesity, increasing age, and sedentary lifestyle are the same as those for VTD 5there is increasing evidence of a hypercoagulable state in OSA.6,7 Some studies have suggested significant associations between OSA and increased fibrinogen and D-dimer6 and increased concentrations of coagulation factors (including thrombin and antithrombin, and von Willebrand factor),8 higher serum levels of tissue factor 8increased platelet activity and aggregability,9,10 impaired fibrinolytic activity 7and whole-blood hypercoagulability. However, hemostasis is a complex process that encompasses blood clotting, platelet activation, and vascular repair, involving multiple molecules and cell types, such as platelets, endothelium cells, and leukocytes; thus, its evaluation requires an integrated examination. Here, we investigated the potential utility of rotational thromboelastometry (ROTEM) and calibrated automated thrombogram (CAT) in the characterization of the procoagulant state in patients with severe OSA and assessed whether these tests offer new insight into the physiopathological mechanisms of OSA and its procoagulant profile.
MethodsNewly diagnosed OSA patients over 35 years of age with an apnea-hypopnea index (AHI)≥30/h on respiratory polygraphy and without previous comorbidities or history of thrombosis were consecutively included. Healthy volunteers without OSA, who were homogeneous in sex, age and smoking habit were selected as controls.
The study was approved by the local Ethics Committee (registry number PI-1857), and informed consent was obtained from all participants. A more detailed description of the methods is provided in the online data supplement.
Platelet-free plasma (PFP) and platelet-rich plasma (PRP) were prepared as explained in the online data supplement.
Rotational thromboelastometry was performed in whole blood by ROTEM Gamma (Pentapharm, Munich, Germany). ROTEM evaluates clotting time (CT: time from start of test until 2mm of amplitude, in seconds); clot formation time (CFT: time from CT to an amplitude of 20mm, in seconds); α angle (tangent to the curve at 20mm amplitude, in degrees); amplitude at “×” time (in mm); maximum clot firmness (MCF: reflects the maximum strength of the clot, in mm); maximum velocity of clot formation (MAXV), and lysis at 60min (LI60, indicates residual clot firmness at 60min, in %).
Surface expression of fibrinogen and von Willebrand factor receptors as well as activation markers (activation of fibrinogen receptor and P-selectin and tetraspanin CD63 release from alpha and dense granules, respectively), and basal and thrombin receptor-activating peptide 6 (TRAP)- and adenosine diphosphate (ADP)-induced leukocyte-platelet aggregates were determined by flow cytometry, as described in online data supplement.
Thrombin generation was assessed in platelet-free plasma (PFP) in fresh by calibrated automated thrombogram (CAT) employing PPP-Reagent LOW. Procoagulant activity associated with microparticle (MP) content of either tissue factor (TF) or phosphatidylserine (PS) was determined using, respectively, MP-reagent and PRP-reagent. The lagtime (time from start of test until 10nM thrombin was formed, in min), the peak height of the curve (the maximum thrombin concentration generated, in nM); the time to reach the peak and endogenous thrombin potential (total amount of thrombin generated over time, in nMxmin) were determined. More details are given in the in online data supplement.
E-selectin and cell free DNA (cfDNA) were quantified, respectively, by ELISA (R&D Systems Europe Ltd., Abingdon, UK) and by a fluorometric test in PFP as mentioned in online data supplement.
Data were analyzed by GraphPad Prism software 5.03. Normal distribution of the data was tested using the Shapiro–Wilk test. Mann–Whitney or Student's t-tests were performed, and data were expressed as median (percentile 25%–percentile 75%) or mean±standard deviation depending on sample distribution.
The correlation analysis was performed using Pearson's or Spearman's test. All tests were two-tailed, and statistical significance was set at p<0.05.
Comparisons between OSA patients and control subjects were adjusted by anthropometric characteristics by linear general model univariate analysis, using group as fixed factor and sex, age, BMI and neck circumference as covariates.
ResultsPatient populationThirty-eight patients with severe OSA and nineteen healthy age- and sex-matched controls were included. The clinical characteristics of the patients are summarized in Table 1. Although patients with OSA had a higher BMI than control subjects, the percentage of obese patients was similar in both groups (51.4 vs. 42.1%, p=0.355).
Baseline characteristics of the study participants.b
| Severe OSA group(n=38) | Healthy volunteers(n=19) | p-Valuea | |
|---|---|---|---|
| Age, years | 60±11 | 58±11 | 0.427 |
| Males, n (%) | 28 (74) | 12 (63) | 0.301 |
| Body mass index, kg/m2 | 31.9±5.2 | 28.6±3.2 | 0.013 |
| Neck circumference, cm | 38 (33–43) | 36 (32–38) | 0.038 |
| Smoking habit, n (%) | 0.928 | ||
| Current smoker | 11 (29) | 5 (26) | |
| Former smoker | 13 (34) | 6 (32) | |
| Never smoker | 14 (37) | 8 (42) | |
| Epworth Somnolence Score (ESS) | 8±5 | 3±2 | <0.001 |
| Daytime sleepiness (ESS≥10) | 13 (34) | 0 | <0.001 |
| AHI, events/h | 48.5±16.5 | 2.8±1.2 | <0.001 |
| Oxygen desaturation index, events/h | 47.0±17.6 | 2.9±0.9 | <0.001 |
| tSpO2<90%, % | 28.4±24.5 | 0.5±0.8 | <0.001 |
| Mean nocturnal SpO2, % | 91±2 | 95±1 | <0.001 |
| Low nocturnal SpO2, % | 75±9 | 89±1 | <0.001 |
| Obstructive events, % | 87.2±6.4 | 86.0±6.4 | 0.343 |
| Systolic blood pressure, mm Hg | 124±10 | 122±10 | 0.359 |
| Diastolic blood pressure, mmHg | 85±7 | 73±6 | 0.421 |
| Red blood cell count×106, per μl | 4.5±0.5 | 4.4±0.5 | 0.560 |
| Hemoglobin, g/dl | 14.1±1.3 | 13.4±1.7 | 0.173 |
| Hct, % | 42.9±4.9 | 42.1±3.5 | 0.339 |
| MCH, pg | 31.2±1.3 | 30.3±1.8 | <0.05 |
| MCHC, g/dl | 33.0±0.6 | 31.2±1.3 | <0.0001 |
| RDW, % | 13.2±0.6 | 15.0±1.8 | <0.0001 |
| White cell count×103, permm3 | |||
| Total | 6.5±1.7 | 6.7±1.7 | 0.333 |
| Neutrophils | 4.0±1.3 | 4.4±1.3 | 0.204 |
| Lymphocytes | 2.1±0.6 | 2.0 (1.8–2.2) | 0.828 |
| Monocytes | 0.3 (0.2–0.5) | 0.5±0.5 | 0.156 |
| Platelet count×103, per μl | 213.6±64.3 | 216 (180–238) | 0.847 |
| MPV, fl | 7.1±0.5 | 7.4±0.7 | 0.116 |
| PDW, % | 17.4±0.8 | 17.3±0.9 | 0.757 |
| Cholesterol total, mg/dl | 186±50 | 173±33 | 0.320 |
| HDL-cholesterol, mg/dl | 124±46 | 113±30 | 0.352 |
| LDL-cholesterol, mg/dl | 51±6 | 50±4 | 0.855 |
| Triglycerides, mg/dl | 151±49 | 129±20 | 0.065 |
Abbreviations: AHI=apnea–hypopnea index; ESS=Epworth somnolence score; HDL=high-density lipoprotein; LDL=low-density lipoprotein; Hct=hematocrit; MCH=mean corpuscular hemoglobin; MCHC=mean corpuscular hemoglobin concentration; MPV=mean platelet volume; PDW=platelet distribution width; RDW=red cell distribution width; SpO2=oxyhemoglobin saturation; tSpO2<90%=time with SpO2<90%.
The ROTEM studies showed significant differences in the dynamics of clot formation when comparing control and OSA samples. Patients with OSA had an increased alpha angle and MAXV, and an augmented clot firmness assessed by the amplitude at 10min and MCF. Nevertheless, CT and clot lysis after 60min was similar in both groups (Fig. 1).
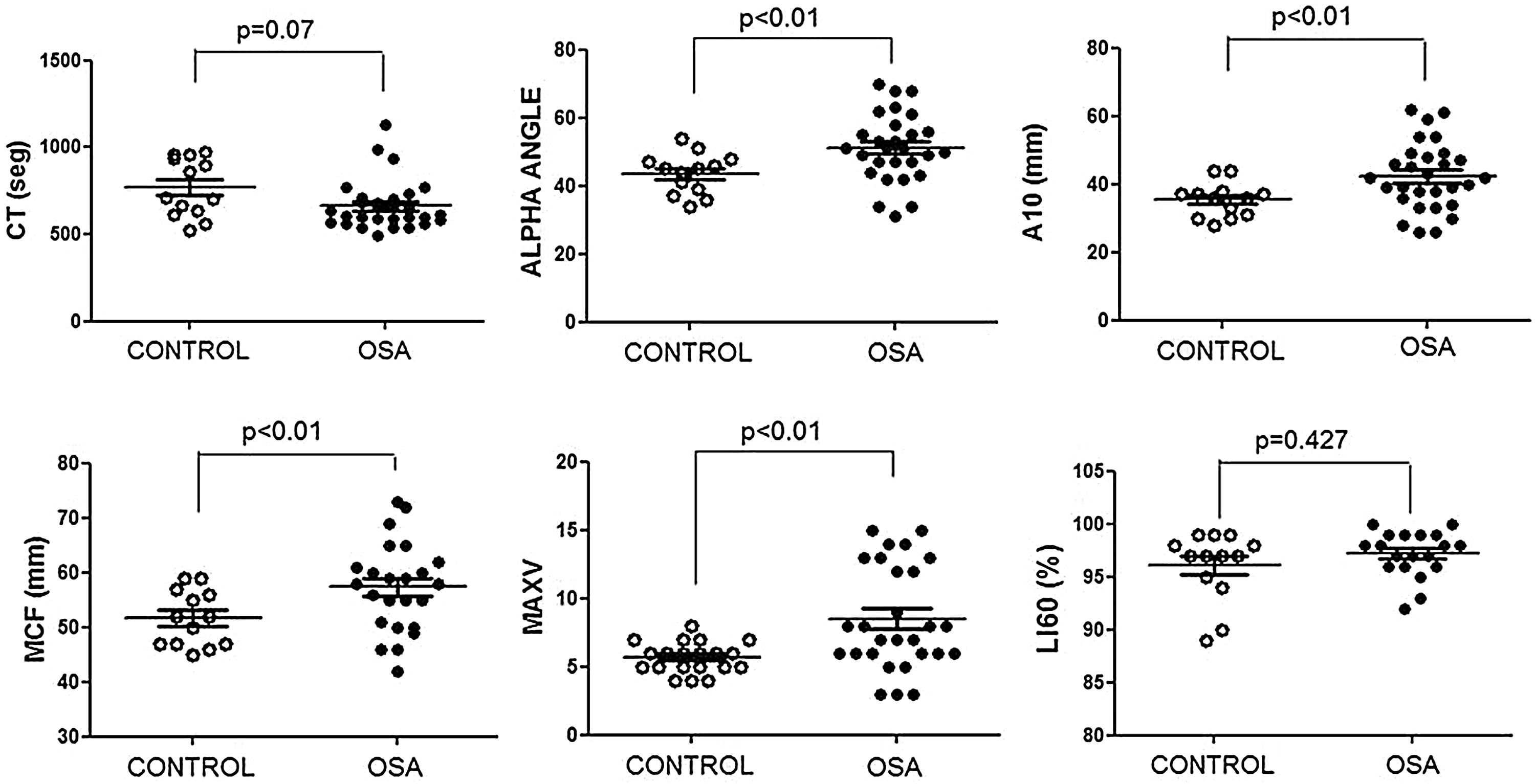
Features of clot formation in patients with severe OSA. Thromboelastometry was performed in whole blood. CT: time from start of test until 2mm of amplitude; α angle tangent to the curve at 20mm amplitude; MCF: maximum clot firmness; MAXV: maximum velocity of clot formation, LI60: clot lysis at 60min. The detailed procedure is explained in the Methods section. Mann–Whitney test (for CT analyses) or Student's t test was performed, and p<0.05 was considered significant.
These results showed procoagulant behavior in the hemostasis of the patients with OSA. This clotting test provided integrated information on blood cells and plasma interactions; thus, we attempted to elucidate the role of each of these elements in this thromboelastogram profile.
Functional state of platelets from patients with obstructive sleep apneaPlatelets from patients with OSA were not activated in basal conditions, and their ability to respond to agonists (as evaluated by the activation capacity of fibrinogen receptor and of the release of granules’ content) was similar to that observed in platelets from healthy controls. Moreover, the surface exposure of receptors for adhesion ligands was the same in both groups (data not shown).
Plasma and microparticle-associated procoagulant activity in patients with obstructive sleep apneaFig. 2A shows the thrombin generation (CAT parameters) obtained with PFP samples from the controls and the patients with OSA. Increased peak of thrombin generation was observed in the patients with OSA, indicating that plasma from these patients had a procoagulant profile independent from the procoagulant capacity of MP.
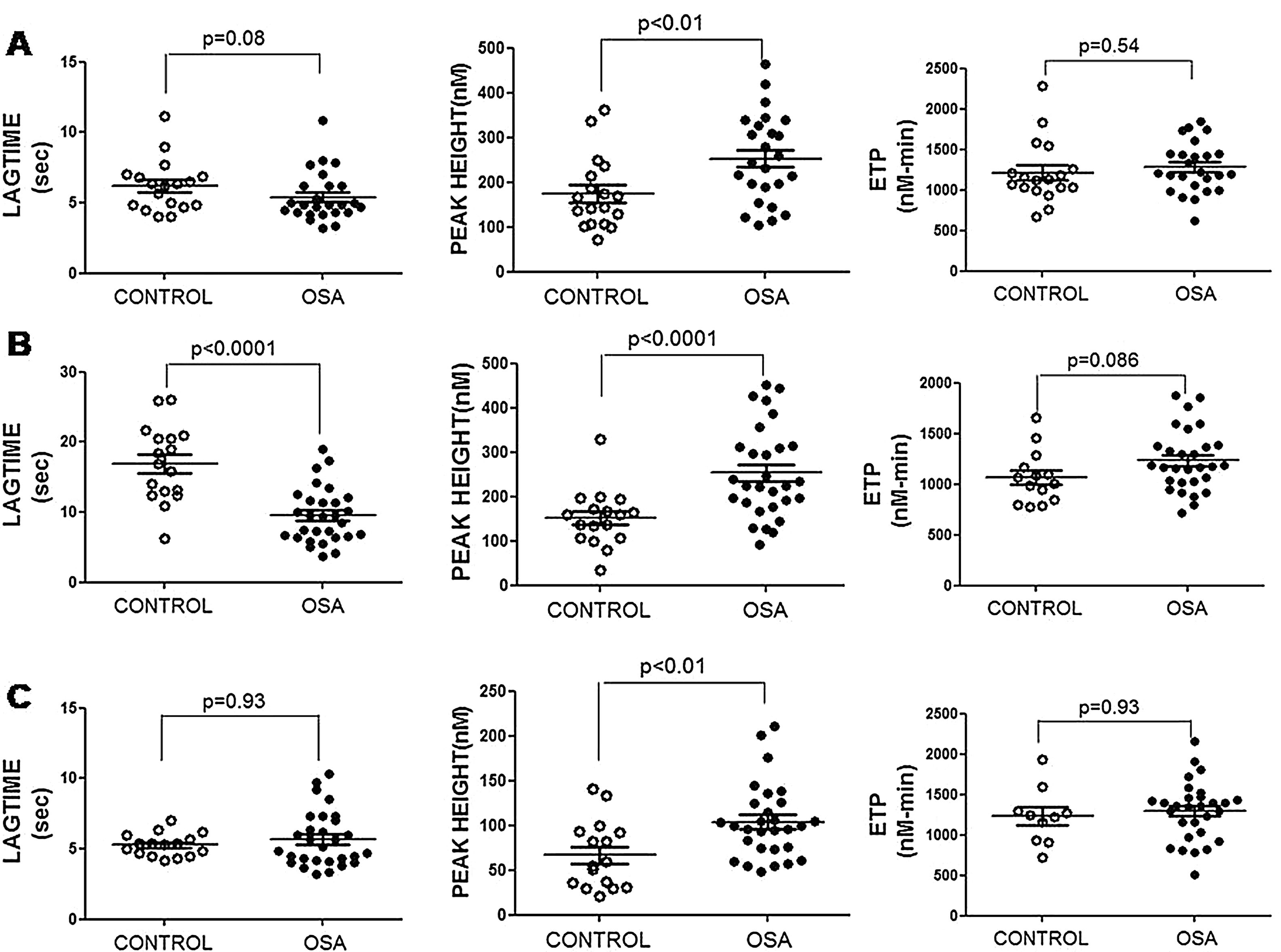
Plasma- and MP-associated procoagulant capacity in patients with severe OSA. Procoagulant capacity was measured by CAT in plasma samples triggered by the following reagents: PPP-Reagent LOW (A), MP-Reagent (B), and PRP-Reagent (C) to evaluate, respectively, plasma, tissue factor (TF)-associated and phosphatidylserine (PS)-associated procoagulant activity of MPs. Mann–Whitney test (for Lagtime-PPP, and Peak height-PRP analyses) or Student's t test was performed, and p<0.05 was considered significant.
Thrombin generation associated with the TF (MP-Reagent) and PS (PRP-Reagent) content of MPs was also determined. As observed in Fig. 2B and C, patients with OSA had increased thrombin generation related to MPs. Moreover, this effect appeared to be predominantly linked to the TF content of MPs.
The plasma content of nucleic acids might contribute to the creation of prothrombotic profiles. We observed that plasma from patients with OSA had increased free nucleic acids (Fig. 3A).
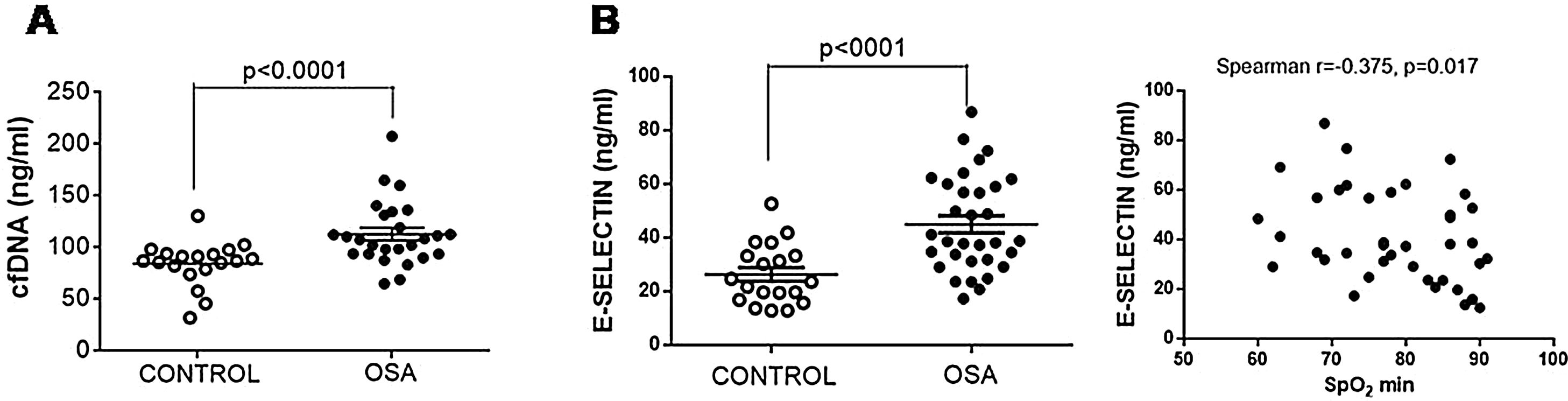
E-selectin and cfDNA in plasma from patients with severe OSA. (A) Free nucleic acids (cfDNA) in plasma by a fluorometric assay. (B) E-selectin was determined in plasma samples by ELISA. The relationship between plasma levels of E-selectin and low nocturnal SpO2 is shown. Mann–Whitney test and Spearman's test were performed, and p<0.05 was considered significant.
Given TF-rich MPs might originate from endothelial cells as a consequence of endothelial damage, plasma concentration of E-selectin, a marker of endothelial injury, was determined. E-selectin was increased in samples from patients with OSA (Fig. 3B). Moreover, E-selectin plasma levels correlated with low nocturnal SpO2 (Fig. 3B).
Formation of leukocyte-platelet aggregates in patients with severe obstructive sleep apneaPatients with OSA formed more leukocyte-platelet aggregates than healthy controls in basal conditions and after stimulation with TRAP and with ADP (Fig. 4).
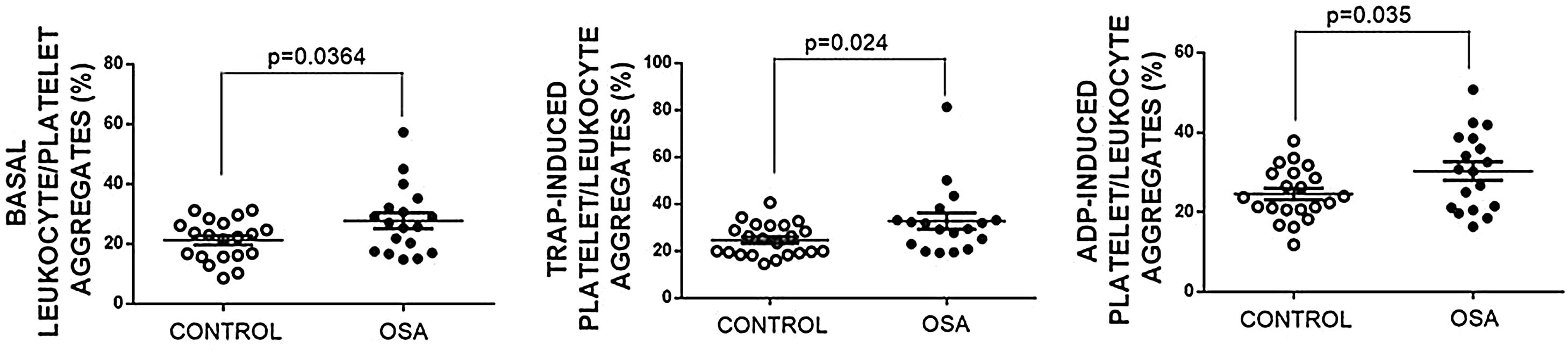
Leukocyte/platelet aggregates in controls and patients with severe OSA. Leukocyte/platelet aggregates in whole blood, in basal conditions, and after stimulation with 20μM TRAP or 10μM ADP. Data are expressed as % of leukocytes positive for αIIb. Mann–Whitney test (for analyses of basal and TRAP-induced aggregates) or Student's t test was performed, and p<0.05 was considered significant.
Enhancement of thrombin generation related to plasma as well as to MPs from patients with OSA appeared to be related to the severity of the disease. Patients with the highest AHI presented the most severe procoagulant profile (Fig. 5A). On the other hand, the minimum mean nocturnal and low nocturnal levels of SpO2 were associated with the highest amount of thrombin generated by plasma (Fig. 5B) and MPs (Fig. 5C).
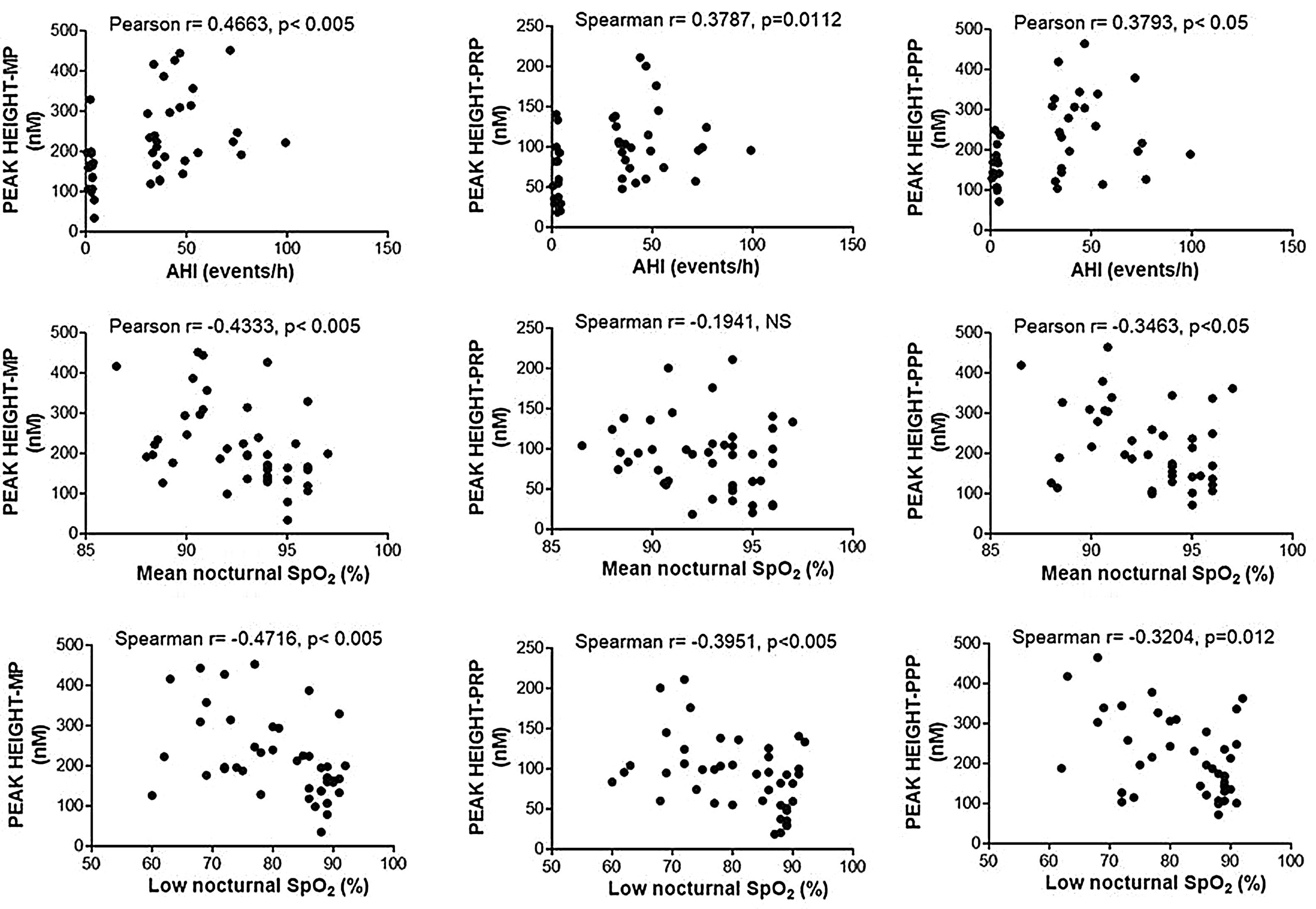
Correlation between clinical parameters and peak of thrombin generation. Thrombin generation was triggered with either MP- or PRP- or PPP-LOW reagents. Correlation between Peak and: (A) apnea-hypopnea index (AHI), (B) mean nocturnal SpO2, and (C) low nocturnal SpO2 are shown. All participants (controls and severe OSA patients) were included in the analysis. Data was analyzed with Spearman's (for analyses of correlations with either Peak height-PRP or Low nocturnal SpO2) or Pearson's test, and p<0.05 was considered significant.
The present study shows that thrombin generation and blood clotting capacity were increased in patients with severe OSA without comorbidities, even in the absence of a previous history of thrombosis. Additionally, we found significant correlations between parameters of thrombin generation and AHI, mean nocturnal SpO2, and low nocturnal SpO2, which suggests links between the procoagulant state, and the severity of sleep disorder.
Hypercoagulable features of OSA patients might be due to several factors that can alter hemostasis. For example, it has been reported that patients with OSA have higher hematocrit values, leading to an increase in blood viscosity that correlates with OSA severity.11 Nevertheless, we did not observe differences in hematocrit values between our cohort of patients with OSA and healthy controls. Moreover, and contrary to Archontogeorgis et al.,12 we observed similar values in platelet distribution width and mean platelet volume (MPV) between control subjects and apneic patients. These findings suggest that neither hematocrit nor platelet width nor MPV contributed to the higher clotting capacity observed in patients with OSA.
Another factor that could influence the kinetics of clot formation is the platelet activation state and count. However, in our cohort of OSA patients, platelets were not activated in quiescent conditions and had a similar ability to be activated by agonists as those from healthy controls. Our results are in contrast with a previous by von Kanel and Dimsdale,13 which analyzed five studies on platelet activity in patients with OSA. Although three studies showed higher platelet activation in these patients compared with controls, only one of them provided the statistical signification of this difference. Moreover, another sign of the lack of basal activity in platelets from our cohort of patients with OSA is that MPV, a marker of increased platelet activity,14 was similar to that observed in the healthy control group.
Regarding the ability of platelets from patients with OSA to be activated by agonists, opposite results have also been reported. Our results and those from West et al.15demonstrated that platelets from OSA patients and healthy controls had similar responses to agonist stimulation. On the other hand, Alkhiary et al. reported an increased ADP-induced aggregation of platelets from these patients.16 However, since a more comprehensive description of their patients is not available, the presence of comorbidities that favor platelet activation cannot be excluded. Even other authors 17have reported that OSA patients have a decreased platelets aggregation after stimulation with epinephrine but not with ADP or TRAP. All these controversial results on platelet activity and ability to be activated might rely on the characteristics of the cohort of patients as well as on the techniques employed for measuring platelet activation.
However, we observed that patients with OSA presented more platelet/leukocyte aggregates in basal conditions even though they did not have basally activated platelets. This situation can be explained because chronic intermittent hypoxia leads to the production of proinflammatory mediators, such as IL-1β, IL-6, and tumor necrosis factor-α,18 and inflammatory stimuli are able to increase the formation of platelet-leukocyte complexes in whole blood 19without inducing platelet aggregation.20 These results support the hypothesis of a dichotomy in platelet activation depending on the inflammatory versus hemostatic nature of the stimulus.21
Other important finding of our study is the identification that plasma from severe OSA patients has increased thrombin generation potential. In contrast, von Kanel et al. found no differences in thrombin–antithrombin and d-dimer (hypercoagulability markers) between patients with OSA and non-OSA controls.22 However, we believe our results are strong because we measured thrombin generation by a direct technique and not through its degradation products.
Increased clot strength in samples from patients with OSA might also rely on an augmented presence of circulating MPs. Although we did not measure levels of MPs by flow cytometry, we can assume they were increased in our cohort of patients because we observed an enhancement in thrombin generation associated with PS- and TF-containing MPs, and a correlation between their functional activity and their number has been reported.23 In fact, it has been reported that MPs from monocytes and endothelial cells are the richest TF-containing MPs. Moreover, elevated level of circulating endothelial MPs have previously been reported in patients with OSA without other comorbidities.24 MPs from monocytes are also considered one of the most thrombogenic MPs.25 They can be captured by activated platelets within thrombi through a P-selectin/P-selectin glycoprotein 1-dependent mechanism that increases fibrin deposition 26and docks on endothelial cells, inducing their activation and apoptosis. Along these lines, El Solh et al. demonstrated that patients with OSA have an enhanced apoptosis of endothelial cells that is strongly correlated with soluble TF.27
We observed that patients with severe OSA had increased levels of cfDNA in plasma, another factor that can induce a procoagulant profile.28 CfDNA is released from necrotic and apoptotic cells such as endothelial cells upon activation by proinflammatory cytokines. So, cfDNA can be considered a good marker of endothelial damage.29 Similar results were observed by other authors.30,31 Elevated plasma levels of E-selectin observed in our cohort of patients confirmed endothelial damage in these patients with OSA. Moreover, we observed that E-selectin levels inversely correlated with nocturnal SpO2, confirming the relationship between hypoxia and endothelial noxa. Moreover, Cofta et al. reported that soluble E-selectin is the most sensitive indicator of cardiovascular risk among other selectins tested, given it progressively increased severity of OSA.32
Correlation analyses of our results have shown associations between AHI, mean nocturnal SpO2, and low nocturnal SpO2 with thrombin generation dependent on either plasma or PS- and TF-associated MPs. These significant associations suggest that procoagulant changes vary with OSA severity. Other authors have reported a relationship between mean nocturnal SpO2 and fibrinogen in patients with OSA,33 providing additional evidence of the involvement of the blood coagulation system in the increased risk of cardiovascular events observed in these patients.
To our knowledge, only two studies have previously used global tests to determine the kinetics of clot formation, but in these cases they did not use ROTEM, but another global test (thromboelastography) to assess coagulation in patients with OSA.34,35 One notable observation was that although patients with OSA reportedly have increased plasma levels of plasminogen activator inhibitor-1 (PAI-1), an inhibitor of fibrinolysis 36we did not detect differences with controls in lysis’ values of ROTEM experiments. Similarly, Toukh et al. observed no changes in this parameter.35 It is tempting to speculate that these tests are not sensitive enough to detect hypofibrinolytic states, but this hypothesis does not appear to be true because we did observe hypofibrinolysis in patients with immune thrombocytopenia.37 Another similarity with Toukh experiments was the increased clot strength observed in the patients with OSA and the lack of effect of OSA on clot formation onset time. On the other hand, Guardiola et al. observed a reduction in this time.34 Differences in these results might be due to differences in the study design. None of these studies included a control group, as ours did. Furthermore, the Toukh study had a crossover design in which each patient served as their own control, whereas Guardiola et al. compared independent groups of patients with OSA.
Our study has several limitations that must be acknowledged. First, it is a small, single-center study focused exclusively on patients with severe OSA, so the results should be extrapolated with caution to other hospitals or levels of severity. Second, although there is no difference in the percentage of subjects with obesity between the two study groups, the BMI of our OSA patients was slightly higher than that of the control subjects, so we cannot categorically exclude any contribution of obesity to the identified findings. Third, our study does not provide any information on the effect of treatment of OSA on coagulation pathways, and therefore no therapeutic recommendations beyond those in place for OSA criteria can be formulated.
ConclusionOur results suggest that OSA induces an increased procoagulant state characterized by an increase in the dynamics of clot formation although without alterations in fibrinolysis. This state appears to be caused by at least two pathological mechanisms. While there does not seem to be a baseline activation of platelets, they do experience a greater formation of platelet-leukocyte aggregates, which seems more dependent on the OSA-induced systemic inflammation than on the activation of hemostatic stimuli. On the other hand, the plasma of patients with severe OSA shows a greater procoagulant activity, with a greater capacity for the generation of thrombin, which seems to depend mainly on the increase in MPs containing TF that are produced as a consequence of the endothelial damage (Fig. 6). Moreover, this study has identified thromboelastometry and thrombin generation assay as useful tools to evaluate the prothrombotic state in these patients. These techniques might have decision-making potential to guide therapy in patients with OSA.
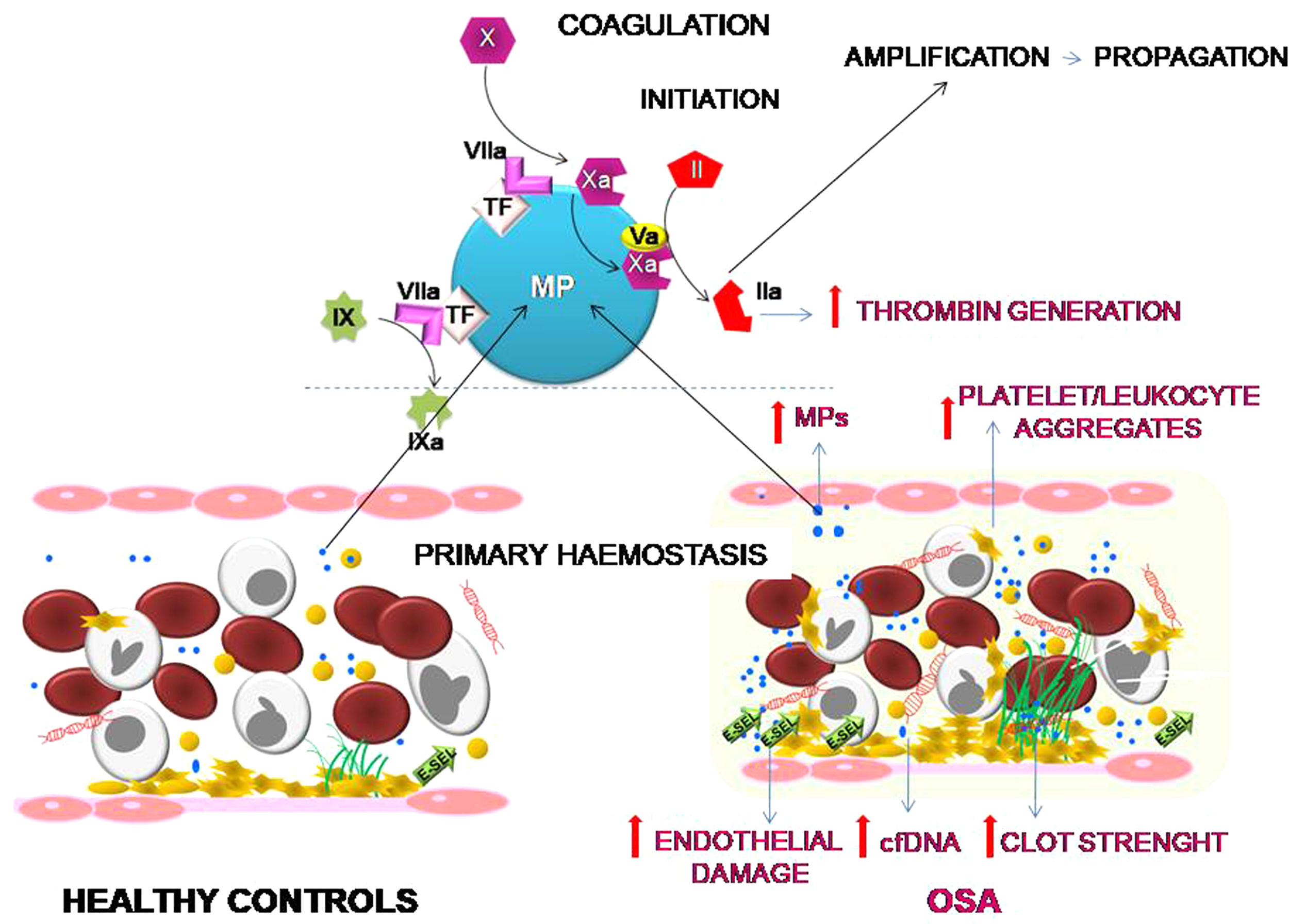
Effects of OSA on primary hemostasis and coagulation. OSA induces the formation of a stronger clot and increased formation of platelet-leukocyte aggregates. Moreover, OSA provokes an endothelial damage that causes a higher release of cfDNA and of MPs containing TF that enhance capacity for the generation of thrombin.
IFB, EMM, RJS, PA and CC-Z performed the experiments; RC, BS, AJ, AA-F and FGR recruited patients and collected clinical data; IFB, MTAR, VJY, FGR and NVB analyzed data of work, FGR and NVB designed the study; NVB wrote the manuscript; all authors revised critically the paper and approved it.
FundingThis work was supported by FIS-Fondos FEDER: PI19/00772 (NVB), PI16/00201 and PI19/01612 (FGR) and PI19/01363 (CC-Z) and PI19/00875 (AA-F). EMM holds a predoctoral fellowship from Fundación Española de Trombosis y Hemostasia (FETH-SETH).
Conflict of interestThe authors declare that they have no conflict of interest.
The following are the supplementary data to this article:





www.publicationethics.org.




Archivos de Bronconeumología follows the Recommendations for the Conduct, Reporting, Editing and Publication of Scholarly Work in Medical Journals