

Introducción
Las infecciones pulmonares, particularmente la neumonía adquirida en la comunidad (NAC), han sido objeto de estudio e investigación desde la antigüedad. Sin embargo, la fisiopatología de esta enfermedad todavía no se conoce con exactitud. En los últimos años se han descrito múltiples factores de riesgo o comorbilidades que se asocian a una mayor probabilidad de muerte por neumonía. Esto ha dado lugar a la aparición de varias escalas pronósticas que han permitido, entre otras cosas, la utilización de un lenguaje homogéneo para calcular la probabilidad de muerte de un paciente con NAC en cualquier lugar del mundo, aunque los mecanismos por los que se produce este aumento del riesgo no siempre están suficientemente claros.
Lo cierto es que los clínicos seguimos observando que las muertes por neumonía también se producen en pacientes sin factores de riesgo ni enfermedades subyacentes; que, si bien la edad es un factor de riesgo, más de la mitad de los fallecimientos por neumonía neumocócica bacteriémica acontecen en pacientes menores de 65 años; que la infección por una cepa idéntica de Streptococcus pneumoniae puede causar shock séptico y muerte en un determinado paciente, o una infección banal y autolimitada en otro. De hecho, es posible que una NAC evolucione mal a pesar de un tratamiento antimicrobiano con espectro adecuado y sensible al microorganismo y, al contrario, tampoco parece haber, al menos en el caso de S. pneumoniae, una asociación clara entre mortalidad y resistencia del microorganismo. Lo que conocemos en la actualidad de la fisiopatología de la neumonía explica muchas de las manifestaciones clínicas específicas que observamos en la práctica, pero no aclara suficientemente por qué sólo algunos pacientes presentan este tipo de manifestaciones o complicaciones.
Mecanismos de defensa frente a la infección
La función principal del pulmón es efectuar el intercambio de gases con la atmósfera. Esta compleja tarea se realiza a través de una interfase alveolocapilar, que constituye la superficie epitelial más extensa del organismo. El aire inspirado, que contiene muchos agentes potencialmente peligrosos, tiene un área de contacto de unos 50-100 m2 con la superficie epitelial del pulmón, lo que, por una parte, facilita la difusión de los gases, pero, por otra, hace que este órgano sea particularmente susceptible a la infección. Como contrapartida, el tracto respiratorio cuenta con numerosos mecanismos de defensa, que comienzan por las barreras anatómicas de la nariz y que se extienden hasta los alvéolos y sus células fagocíticas.
Barreras anatómicas y defensa innata
Cuando se respira por la nariz, las vibrisas nasales son capaces de eliminar partículas mayores de 10-15 μm. En las vías aéreas superiores, las amígdalas y adenoides representan áreas de tejido linfoide secundario y son zonas especialmente dotadas para la eliminación de sustancias extrañas debido a su gran población de leucocitos residentes. Las partículas inferiores a 10 μm alcanzan las vías aéreas inferiores, donde disminuyen las posibilidades de impactación, pero aumentan las de sedimentación en la mucosa. La capa de moco que tapiza los bronquios contiene, entre otras sustancias, unas glucoproteínas, denominadas mucinas, que son capaces de unirse a los microorganismos y neutralizarlos. Además de este efecto directo de las mucinas, las secreciones bronquiales facilitan la eliminación de partículas a través del sistema mucociliar. Las partículas de alrededor de 4 μm de diámetro tienen más probabilidades de alcanzar los alvéolos, dado que son lo bastante grandes para evitar ser exhaladas y lo bastante pequeñas para eludir la impactación precoz en la mucosa de la vía aérea1.
Las bacterias tienen un tamaño óptimo (1-5 μm) para alcanzar los alvéolos. Por consiguiente, tiene que haber otros mecanismos, además de las barreras anatómicas que hemos mencionado, para mantener la esterilidad del pulmón. De hecho, hay múltiples sustancias antimicrobianas (como las defensinas, la lisozima, la lactoferrina, el sistema del complemento, la fibronectina, las inmunoglobulinas y las colectinas) con propiedades bactericidas que facilitan, directa o indirectamente, la eliminación de los microorganismos2.
Reconocimiento del patógeno
Las barreras anatómicas y los péptidos antimicrobianos de las vías aéreas constituyen los elementos constitutivos de la defensa pulmonar en el huésped sano, siempre listos para responder a la presencia de microorganismos. Sin embargo, si el inóculo bacteriano que se aspira es importante o está constituido por organismos encapsulados más virulentos, estos factores defensivos no siempre son suficientes para evitar la infección bacteriana, y la probabilidad de que los patógenos lleguen a las zonas más distales del pulmón y proliferen de forma incontrolada es mayor. En estas circunstancias son necesarios mecanismos defensivos adicionales para impedir el desarrollo de una neumonía. La eliminación de los microorganismos que alcanzan el alvéolo depende, básicamente, de los macrófagos alveolares, que constituyen la primera línea defensiva celular pulmonar en los sujetos sanos. El inicio de una respuesta inmunitaria, orquestada por el macrófago alveolar, requiere el reconocimiento del patógeno y una clara distinción entre lo "propio" y lo "extraño".
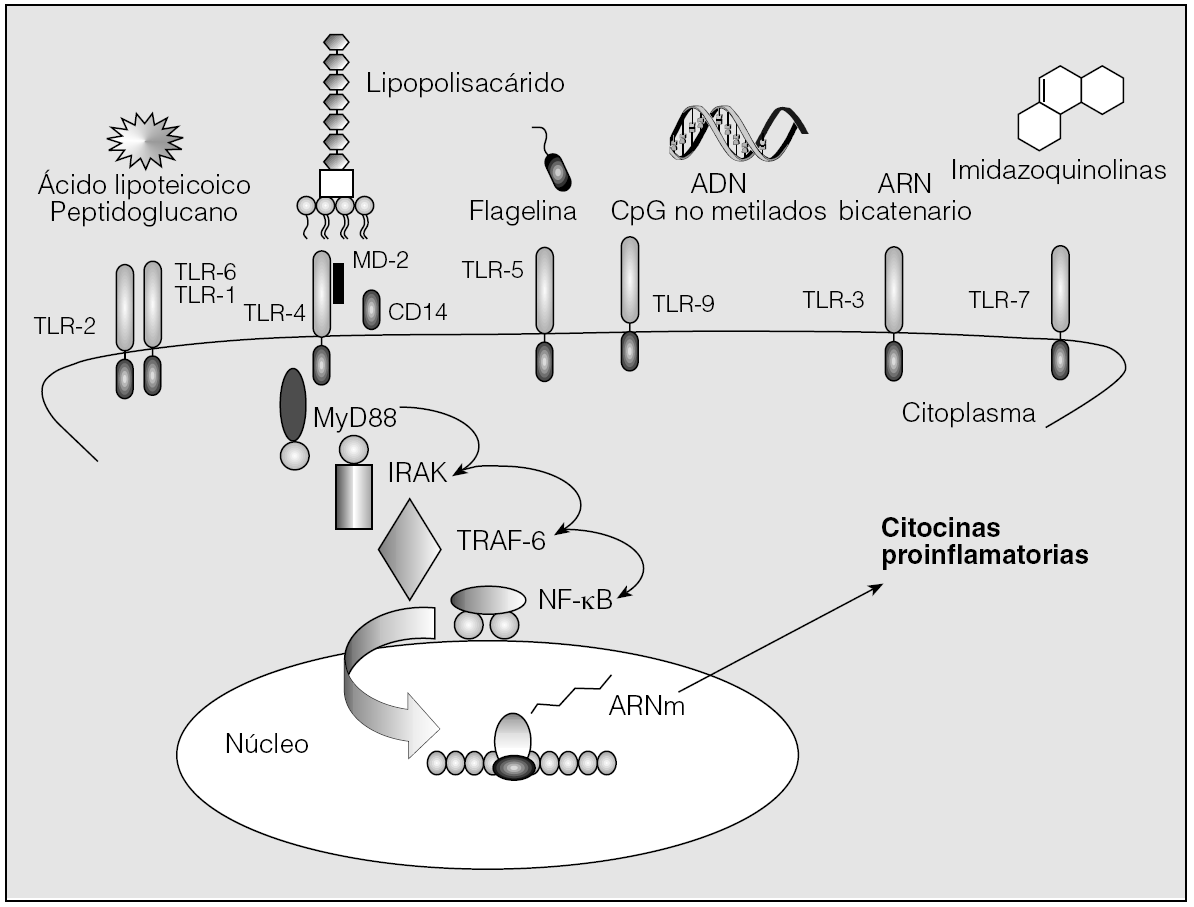
A lo largo de la evolución, la inmunidad innata ha desarrollado un sistema muy eficaz de reconocimiento de un patrón molecular común y constante de la superficie de los microorganismos denominado patrón molecular asociado a patógenos (PMAP), a través de los llamados receptores reconocedores de patrones (RRP). Los PMAP son característicos de los microorganismos, lo que permite al sistema inmunitario innato distinguir entre antígenos propios y extraños; son invariables, de forma que con un número limitado de RRP se detecta la presencia de cualquier patógeno; y son esenciales para la supervivencia o patogenidad del microorganismo, por lo que sus mutaciones son letales. Entre los principales PMAP, que actúan como dianas para la activación del sistema inmunitario innato, se encuentran productos de la fisiología microbiana como el lipopolisacárido (LPS) (gramnegativos), ácido lipoteicoico y peptidoglicano (grampositivos), lipoarabinomán (micobacterias), cimosan (levaduras), secuencias de ADN con dominios CpG no metilados, manosa o ARN bicatenario (virus). Por otra parte, hay distintos tipos de proteínas que son capaces de reconocer PMAP. Entre estos RRP se encuentran proteínas del sistema del complemento, como la lectina de unión a manosa, receptores endocíticos, como los receptores de la manosa; y, por último, receptores de membrana, como los "receptores tipo toll" (TLR) y CD14 (fig. 1), que se expresan fundamentalmente en la superficie de las células que primero entran en contacto con el patógeno durante la infección (células de la superficie epitelial) y en las células presentadoras de antígenos (monocitos/macrófagos y células dendríticas)3,4.

Fig. 1. Receptores tipo toll(TLR) y vías de señalización celular en la respuesta inflamatoria. ARNm: ARN mensajero; IRAK: cinasa asociada al receptor de interleucina-1; MyD88: factor de diferenciación mieloide-88; NK-κB: factor nuclear-κB; TRAF-6: factor asociado al receptor de necrosis tumoral alfa.
El reconocimiento de la bacteria por los macrófagos alveolares es un proceso complejo y fundamental para el inicio, la expansión, el mantenimiento y la resolución de la respuesta defensiva del huésped frente a la infección pulmonar. Al unirse los PMAP a los RRP se inicia una cascada de señalización intracelular y comienza una serie de procesos antimicrobianos y funciones defensivas. Básicamente, lo que sucede es que se activa el factor de transcripción nuclear-κB, se transloca al núcleo celular y se une a la región del promotor, provocando la transcripción de mediadores proinflamatorios factor de necrosis tumoral alfa (TNF-α), interleucina (IL) 1β, IL-6 e interferón-gamma (IFN-γ) y antiinflamatorios IL-10 y el factor transformador de crecimiento-β, que son los reguladores proteicos clave de la inflamación, orquestados por el macrófago alveolar y dirigidos a la eliminación de los patógenos del aparato respiratorio inferior5 (fig. 1). Comentar la respuesta inflamatoria frente a la infección sin la concomitante reacción antiinflamatoria es difícil. Las respuestas están ligadas y las relaciones entre ellas, cuando hay una respuesta apropiada o cuando se da una respuesta patoló-gica inapropiada, son dependientes del tiempo. Una respuesta inicial proinflamatoria insuficiente puede provocar la muerte por infección incontrolable. Por el contrario, una respuesta proinflamatoria excesiva y prolongada puede inducir el fracaso del órgano afectado (pulmón), diseminar la respuesta y provocar un fracaso multiorgánico.
Respuesta inflamatoria
Si la actividad macrofágica está alterada o es insuficiente, se requiere el concurso de los leucocitos polimorfonucleares (PMN) para controlar la proliferación de gérmenes en el espacio alveolar. Estas células no forman parte de la población normal del alvéolo, pero constituyen la población más abundante de fagocitos circulantes, y pueden reclutarse desde el compartimiento vascular gracias a la acción quimiotáctica de las citocinas liberadas tanto por los microorganismos como por los macrófagos activados. Entre estas citocinas se encuentran el TNF-α y la IL-1β, cuyos efectos autocrinos y paracrinos activan los leucocitos, incrementan tanto la permeabilidad vascular como la expresión de moléculas de adhesión y estimulan la producción de nuevas citocinas y quimiocinas2.
Los neutrófilos humanos tienen un tamaño similar al de los hematíes, pero son mucho menos deformables. Por este motivo, estas células tardan hasta 120 s en atravesar un capilar pulmonar, mientras que un glóbulo rojo lo hace en menos de un segundo. Estas circunstancias determinan un secuestro temporal de PMN en el pulmón y, como consecuencia de ello, su concentración en el lecho vascular pulmonar es 100 veces mayor que en la circulación extrapulmonar. Además, debido a su lento tránsito a través de los pulmones, este agrupamiento de neutrófilos marginados es capaz de responder de forma inmediata a estímulos infecciosos dentro del espacio alveolar. La transmigración de los PMN desde el lecho sanguíneo al espacio alveolar supone la expresión coordinada de moléculas de adhesión y de quimioatrayentes, y algunos reajustes en el citosqueleto de estas células.
La marginación y el rodamiento de los neutrófilos sobre el endotelio vascular se inician por la expresión de selectinas. La L-selectina, que se expresa de forma constitutiva en la superficie de los PMN, se une a sialomucinas de las células endoteliales e induce la marginación de los neutrófilos en la vasculatura pulmonar. En condiciones normales los neutrófilos pasan a través de la circulación pulmonar y, aunque lentamente, terminan incorporándose a la circulación sistémica. Sin embargo, cuando hay una infección pulmonar, los mediadores inflamatorios generados en el espacio alveolar regulan al alza la expresión de múltiples moléculas de adhesión en las células endoteliales, incluidas la E-selectina, la P-selectina y la molécula de adhesión intracelular-1. Estas moléculas de adhesión se unen a sus respectivos ligandos en la superficie de los neutrófilos marginados e incrementan su adherencia al endotelio vascular. Además de las selectinas mencionadas, también aparecen quimiocinas de origen pulmonar, como la IL-8, que son claves en la adhesión de los PMN al endotelio y en su posterior migración al tejido inflamado. La interacción de selectinas y quimiocinas con sus respectivos ligandos desencadena vías de señalización transmembrana dentro de los neutrófilos que conducen a la expresión de integrinas-β2 CD11/CD18 y a la liberación de L-selectina. La adherencia firme de los neutrófilos a la superficie endotelial se establece a través de la unión de las integrinas con las moléculas de adhesión intracelular-1 y 2, y dependerá del estímulo inflamatorio. La forma en que los neutrófilos son marginados en el pulmón es diferente de la que ocurre en el resto del organismo.
Una vez secuestrados en la microvasculatura pulmonar y firmemente adheridos a las células endoteliales, los PMN se aplanan y se desplazan a través de las uniones intercelulares en un proceso conocido como quimiotaxis. La migración directa hacia los alvéolos infectados depende de la expresión de quimioatrayentes de neutrófilos liberados durante la inflamación tanto por el huésped como por el microorganismo. Entre estos quimioatrayentes están el leucotrieno B4, C5a, el factor de activación de las plaquetas y, especialmente, las quimiocinas, cuyo principal representante en humanos es la IL-8. A medida que los neutrófilos migran y se unen a los quimioatrayentes, se inducen múltiples vías de señalización intracelular que convergen característicamente en la activación de MAPK (proteincinasa activada por mitógeno), que da lugar a modificaciones en la configuración citosquelética y a la aparición de lamelipodias (fibras de estrés), que permiten al neutrófilo migrar hacia el estímulo quimiotáctico6,7.
En el espacio alveolar, los neutrófilos se convierten en la principal población fagocítica y destruyen a los microorganismos mediante la generación de oxidantes tóxicos en el fagosoma. El equipo enzimático responsable de este proceso es la nicotinamida adenindinucleótido fosfato (NADPH) oxidasa asociada a la membrana del fagosoma, que en la denominada explosión o estallido respiratorio (respiratory burst) toma electrones de la NADPH para reducir oxígeno a O2. La destrucción de las bacterias por parte de los PMN implica, por tanto, la fagocitosis de aquéllas, la explosión respiratoria y la producción mantenida de péptidos proinflamatorios, todo lo cual se ve favorecido por opsoninas, citocinas y otros factores presentes en el seno de la inflamación. Es evidente que la existencia de defectos en el reclutamiento de PMN se asociará a una mayor dificultad en la destrucción de las bacterias y a un aumento de la mortalidad.
Regulación de la respuesta inflamatoria
La magnitud de la respuesta inflamatoria frente a la infección debe ser proporcionada y permanecer adecuadamente compartimentada para evitar el daño tisular y los efectos sistémicos de la misma, como son la disfunción miocárdica, la hipotensión, la hipoperfusión de órganos vitales y la acidosis láctica. En general, las concentraciones plasmáticas de los mediadores inflamatorios parecen relacionarse con la gravedad de la neumonía o el desarrollo de sepsis, y de hecho la progresión de la neumonía puede producirse por el desequilibrio de la respuesta inflamatoria, incluso en presencia de antibioterapia apropiada. Se desconocen las causas exactas que determinan una respuesta inflamatoria excesiva, aunque es probable que en ella estén implicados varios factores relacionados con el microorganismo causante de la infección y determinadas características del huésped.
Factores microbianos relacionados con la intensidad y naturaleza de la respuesta inflamatoria en la neumonía
Hay una serie de factores directamente relacionados con el patógeno causante de la infección, tales como la proteína tipo III de Pseudomonas aeruginosa8 o la pared celular del neumococo9, que determinan su capacidad invasiva, el daño tisular y la repuesta inflamatoria sistémica. Los cambios citopáticos inducidos por estos factores microbianos pueden determinar una pérdida de la integridad del epitelio o del endotelio alveolar y la incapacidad para compartimentar la inflamación. El LPS o endotoxina de los gramnegativos, probablemente la toxina bacteriana mejor estudiada, es un potente activador del sistema inmunológico. Las bacterias gramnegativas difieren entre sí en la cantidad de LPS que liberan espontáneamente en el curso de una infección. Así, por ejemplo, Neisseria meningitidis es capaz de liberar grandes cantidades de endotoxina y condicionar shock y el daño multiorgánico característico de la meningococemia. La configuración bioquímica del LPS bacteriano influye también en la magnitud de la inflamación sistémica, en particular el número y longitud de las cadenas acil y el grado de fosforilación de la columna de disacáridos del lípido A10. Así, por ejemplo, el LPS de Escherichia coli tiene mayor bioactividad que el LPS de Klebsiella pneumoniae, cuyas 6 cadenas acil son más largas y tienen una configuración de la columna de disacáridos diferente; por el contrario, las moléculas de lípido A de P. aeruginosa y Chlamydophila trachomatis tienen solamente 5 cadenas acil, que difieren en longitud de las de E. coli y poseen menor actividad estimuladora. Por otra parte, ciertos antimicrobianos, particularmente los betalactámicos, inducen una mayor liberación de endotoxina debido a su rápida capacidad bactericida y, como consecuencia, provocan una mayor respuesta inflamatoria. El significado clínico de este efecto producido por los betalactámicos, especialmente en el caso de las neumonías, no está del todo claro. La producción de toxina es otro aspecto importante relacionado con la virulencia de Staphylococcus aureus. Recientemente se ha descrito la leucocidina de Panton-Valentine como un potente mediador de la inflamación, que además puede destruir leucocitos y contribuir así a la necrosis tisular observada en algunas infecciones producidas por este patógeno11. Por último, hay otros promotores de virulencia del microorganismo que ayudan a eludir los mecanismos de defensa del huésped y facilitan su diseminación y la respuesta inflamatoria sistémica12.
Factores del huésped relacionados con la intensidad y naturaleza de la respuesta inflamatoria en la neumonía
El riesgo de muerte por infección se hereda. Las evidencias más contundentes proceden del estudio clásico de Sorensen et al13, publicado en The New England Journal of Medicine en 1988. Estos autores analizaron las causas de muerte prematura en 1.000 familias con niños adoptados. Utilizando el Registro Danés de Adopciones, llevaron a cabo un estudio de cohortes de los sujetos nacidos entre 1924 y 1926 que fueron adoptados a una edad muy temprana. Siguieron a esta cohorte hasta 1982, momento en el que analizaron la causa de muerte de los sujetos que habían fallecido hasta entonces, centrándose básicamente en las causas de origen infeccioso, cardiovascular o cerebrovascular y neoplásico. Los autores examinaron cuál había sido la causa de muerte de los padres biológicos y comprobaron que, si éstos habían muerto por una infección antes de cumplir 50 años, su hijo tenía un riesgo relativo de fallecer por infección de 5,81. Sin embargo, el hecho de que el padre biológico hubiera fallecido por una neoplasia no confería un mayor riesgo al hijo de morir por cáncer. Por otra parte, el fallecimiento de los padres adoptivos por una infección no confería un mayor riesgo al hijo adoptado de fallecer por esta causa, mientras que si el padre adoptivo moría por cáncer, el adoptado tenía un riesgo 5,16 veces mayor de morir por este motivo. Por tanto, la conclusión que se obtiene de este estudio es que la susceptibilidad y la respuesta a la infección tienen una influencia genética sorprendentemente importante, mientras que el desarrollo de cáncer tiene una notable influencia ambiental. Aunque este estudio constituye una evidencia importante de la base genética de la mortalidad asociada a las enfermedades infecciosas, las causas o los mecanismos de tales muertes no quedan establecidos.
Las enfermedades genéticas clásicas que afectan a la inmunidad del huésped (síndrome de inmotilidad ciliar, hipogammaglobulinemias, deficiencias en el complemento y otras enfermedades monogénicas) no pueden explicar la mayor parte de los efectos que describen Sorensen et al en su estudio, o la elevada mortalidad por neumonía que continúa observándose en los países desarrollados. La incidencia de esta enfermedad invita a pensar que la variante genética que está asociada a esta infección debe de ser relativamente frecuente (un polimorfismo), más que una mutación rara. Por otra parte, el riesgo genético en la neumonía es indudablemente poligénico más que la consecuencia de mutaciones en un solo gen, y la interacción entre distintos genes y entre éstos y los factores ambientales tiene un papel fundamental. Cada factor de riesgo genético probablemente explica sólo un componente del riesgo global, una manifestación específica o una fracción de la población total afectada. Sin embargo, la influencia genética sobre la NAC no se limita a un mayor riesgo de muerte por esta causa. Determinados genotipos pueden afectar a la predisposición para desarrollar una NAC, a la incidencia de manifestaciones específicas de ésta o a la gravedad del cuadro clínico. Hay una gran probabilidad de que la misma variante genética pueda tener un efecto beneficioso en un aspecto de la patogenia como, por ejemplo, una menor predisposición a tener una neumonía y, a su vez, incrementar el riesgo de complicaciones o de mayor gravedad si la neumonía se desarrolla. Las variantes alélicas en la respuesta inflamatoria son las que más frecuentemente tienen estos efectos duales. Por ejemplo, una mayor respuesta del TNF puede evitar el desarrollo de neumonía, pero también incrementar el riesgo de desarrollar síndrome de distrés respiratorio agudo o shock séptico si la neumonía se produce. Todavía encontramos más confusión cuando observamos que un mismo polimorfismo puede tener distinta respuesta frente a antígenos diferentes (grampositivos o gramnegativos). Si a esto le sumamos las dificultades en el diseño de los estudios de asociación genética, la controversia reinante respecto al papel exacto que desempeñan determinados polimorfismos en la respuesta inflamatoria frente a la infección es comprensible. A pesar de ello, lo que parece incontrovertible es que la variabilidad genética es importante en esta respuesta.
Implicaciones clínicas
Como ya se ha mencionado, la primera línea de defensa frente a la invasión y diseminación de los patógenos implica el reconocimiento del microorganismo invasor, su eliminación y una respuesta inflamatoria y antiinflamatoria que sea capaz de restaurar el equilibrio homeostático. Cada uno de estos procesos puede verse afectado por los polimorfismos de los genes implicados, que pueden provocar una mayor susceptibilidad o una mayor resistencia frente a la infección (tabla I). No obstante, el escenario clínico más probable es el de la existencia de múltiples mutaciones, cada una de las cuales tendría efectos modestos en la producción de las moléculas implicadas o en su función, pero que en conjunto condicionarían una mayor repercusión en la defensa frente a la infección3. Se han descrito polimorfismos de genes implicados en el reconocimiento antigénico (proteínas ligadoras de LPS, CD14, TLR, lectina de unión a manosa); en la respuesta inflamatoria (TNF-α, proteína del shock térmico, IL-1 e IL-6), y en la respuesta antiinflamatoria (IL-10, antagonista del receptor de la IL-1), entre otros. Aunque los resultados obtenidos en la mayoría de los estudios no prueban de forma irrefutable el papel o la función de un gen en la patogenia de la infección respiratoria, permiten generar nuevas hipótesis, sugieren nuevos genes candidatos sobre la base de su papel en la respuesta inflamatoria y proporcionan el primer paso en la comprensión de los factores genéticos subyacentes. El empleo de micromatrices (arrays) proporcionará, sin duda, nuevos datos sobre moléculas cuyo papel en la patogenia de la NAC ha pasado inadvertido hasta ahora. En cualquier caso, la posibilidad de identificar características del huésped que predispongan a ciertos individuos a una mayor susceptibilidad o a una respuesta más grave frente a la infección, continúa siendo un objetivo clínicamente importante14. Entretanto, parecería útil disponer de marcadores biológicos capaces de evaluar la respuesta inflamatoria, así como de tratamientos capaces de modularla.

Implicaciones diagnósticas y pronósticas
A finales de la década de 1980, tras la descripción y clonación del TNF-α, la IL-1 y la IL-6, se produjo una verdadera eclosión de trabajos de investigación sobre estas moléculas. La determinación de citocinas es compleja, en parte debido a su corta vida media plasmática y a la presencia de factores bloqueantes, y en parte porque es un procedimiento caro y no disponible de forma generalizada. Actualmente, la utilidad de la medición de las concentraciones séricas de citocinas en pacientes con neumonía es muy limitada. Sin embargo, la detección y cuantificación de IL-6 en plasma, tanto en roedores como en humanos, es relativamente sencilla, lo que ha contribuido a que esta molécula se convierta en uno de los mejores indicadores de la gravedad de la respuesta inflamatoria en pacientes críticos15. De hecho, diversos ensayos clínicos en sepsis grave han empleado las concentraciones de IL-6 como criterio para la administración de agentes anti-TNF-α16. No obstante, como es obvio, correlación no es sinónimo de causalidad, y actualmente persiste la duda de si los títulos plasmáticos de IL-6 contribuyen al fracaso orgánico y a la mortalidad de la sepsis, los evita o simplemente los describe. A fecha de hoy, la literatura médica al respecto permitiría concluir tanto que la respuesta endógena de la IL-6 carece de papel en la evolución de la infección como que mejora la supervivencia o que incrementa la mortalidad. Se han empleado anticuerpos neutralizantes anti-IL-6 en modelos de sepsis y también aquí se han obtenido resultados contradictorios17.
Los reactantes de fase aguda son un grupo heterogéneo de proteínas que se sintetizan predominantemente en el hígado y cuyas concentraciones plasmáticas aumentan rápidamente en presencia de inflamación y de necrosis tisular. Las citocinas que se producen durante el proceso inflamatorio son los principales estimuladores de la producción de estos reactantes de fase aguda. La proteína C reactiva (PCR), cuya denominación procede de su capacidad de reacción con el polisacárido C del neumococo observada en sujetos con neumonía por este organismo, fue identificada en 1930 y, posteriormente, considerada un marcador precoz, sensible e inespecífico de inflamación. El principal estimulador de su síntesis es la IL-6. Se han descrito cambios en las concentraciones de PCR en múltiples entidades nosológicas, como el infarto agudo de miocardio, el asma o la insuficiencia cardíaca, y también en sujetos sanos tras ejercicio intenso y en los períodos puerperal y postoperatorio. Paradójicamente, a pesar del origen de su descripción inicial, su empleo no ha sido muy habitual en pacientes con infección respiratoria, y sólo en los últimos años, tras la comercialización de sistemas automatizados para su determinación, parece haber renacido el interés por el valor diagnóstico y pronóstico de esta molécula en la neumonía. Algunas publicaciones recientes reconocen la utilidad de la PCR en la predicción etiológica y en la estratificación del riesgo de la infección18. La PCR también puede tener un papel en el seguimien-to de esta enfermedad. El 99% de lo adultos jóvenes presentan una concentración media de PCR menor de 10 mg/l. Tras un estímulo adecuado, estos valores pueden aumentar hasta 10.000 veces. Su síntesis comienza muy rápidamente y las concentraciones séricas aumentan a un ritmo superior a 5 mg/dl en las primeras 6 h, para alcanzar su máximo a las 48 h. La vida media plasmática es de aproximadamente 19 h y, cuando el evento que motivó el incremento de su síntesis desaparece, la concentración circulante de PCR desciende rápidamente19. Esta rápida aparición y desaparición hace de esta molécula un marcador interesante en el control de la evolución de la neumonía y en la predicción de su respuesta terapéutica.
La procalcitonina (PCT) es un precursor polipeptídico de la hormona calcitonina. Inicialmente su estructura está constituida por 116 aminoácidos, que tras la acción de diversas dispeptidasas, dan lugar a 3 moléculas: la calcitonina, la catacalcina (carboxipéptido de calcitonina-I) y el extremo N-terminal (aminoprocalcitonina). La infección bacteriana provoca la expresión ubicua del gen CALC-I y una liberación constitutiva de PCT, de forma que durante la sepsis todo el organismo puede contemplarse como una glándula endocrina20. La PCT es inducida durante las infecciones directamente por toxinas microbianas (p. ej., endotoxina) y/o indirectamente por factores humorales (p. ej., TNF-α, IL-1β e IL-6) y la respuesta inmunitaria celular. Esta inducción de su síntesis puede atenuarse por citocinas liberadas durante las infecciones víricas (p. ej., IFN-γ)20,21. Aunque su función biológica no está clara, es importante destacar que la PCT es un mediador clave en la infección sistémica y que en diversos modelos animales se ha observado que su neutralización inmunitaria mejora la supervivencia, lo que indicaría que esta molécula podría ser una buena diana terapéutica22. La síntesis de PCT se produce de forma más rápida que la de otros reactantes de fase aguda (en las primeras 3 h), para alcanzar su pico a las 6 h tras el estímulo infeccioso23. Diversos estudios han establecido su mayor fiabilidad diagnóstica en relación con otros parámetros de diagnóstico de la sepsis, independientemente del origen de la infección24. Al contrario de lo que ocurre con la PCR, las concentraciones de PCT no se ven influidas por el tratamiento con glucocorticoides u otros fármacos inmunodepresores, y tienen un mejor valor predictivo negativo20. Su valor en la orientación terapéutica también ha quedado de manifiesto en un estudio reciente en el que se empleó la PCT como elemento decisivo para la indicación o no de tratamiento antibiótico en la infección respiratoria. Los autores concluyen que la utilización de la PCT permite evitar, de forma segura, tratamientos antibióticos innecesarios y que esta reducción del uso de antimicrobianos es particularmente evidente en pacientes con bronquitis aguda o exacerbaciones agudas de bronquitis crónicas25. En el contexto de la NAC, el empleo de antibióticos en función de las concentraciones de PCT permite una reducción significativa de la duración del tratamiento, pasando de una mediana de 12 a 5 días26. En la neumonía asociada al ventilador se ha comprobado asimismo el valor pronóstico de la PCT27. En otro estudio reciente sobre una población heterogénea de pacientes graves se ha comprobado que las concentraciones máximas de PCT y su incremento diario son predictores de mortalidad por cualquier causa a los 90 días, mientras que la PCR o el recuento leucocitario no lo son28. Por otra parte, es preciso recordar que la PCT también se eleva en enfermedades no infecciosas que condicionan una respuesta inflamatoria sistémica. Además, en pacientes con infecciones localizadas (p. ej., empiema capsulado) los títulos de PCT son habitualmente inferiores a los observados en pacientes con infecciones generalizadas o con hemocultivos positivos. En conjunto, la fiabilidad diagnóstica de la PCT puede compararse con la del péptido natriurético B en el diagnóstico de la insuficiencia cardíaca20.
Implicaciones terapéuticas
Como ya se ha mencionado, la evolución de la respuesta inflamatoria depende de varios factores, incluidas la patogenidad y la duración del estímulo, y también del equilibrio entre respuesta inflamatoria y antiinflamatoria. Dado que una respuesta inflamatoria excesiva, especialmente si es sistémica, puede perpetuar el daño tisular, parece lógico intentar atenuar su magnitud mediante una intervención terapéutica. Los glucocorticoides son los inhibidores naturales más importantes de la inflamación. Inducen una reducción de las concentraciones de las citocinas inflamatorias TNF-α, IL-1β, IL-6 e IL-8, y participan en la activación y translocación nuclear del factor nuclear-κB, que, como ya se ha comentado, es un factor necesario para la máxima transcripción de un número importante de moléculas asociadas a la respuesta inflamatoria frente a la infección. En este sentido, se han realizado diversos ensayos clínicos encaminados a evaluar el efecto de los glucocorticoides en la evolución de la neumonía grave29. En uno de ellos se comprobó una reducción de la respuesta inflamatoria, tanto pulmonar como sistémica, y una mejoría sintomática en los pacientes con neumonía grave tratados con glucocorticoides en comparación con los controles30. Sin embargo, la mayoría de estos estudios no han podido demostrar la superioridad del tratamiento antiinflamatorio en relación con la supervivencia, la aparición de complicaciones, el desarrollo de insuficiencia respiratoria o la estancia hospitalaria. Más recientemente Confalonieri et al31 han apuntado que el tratamiento de la NAC grave con hidrocortisona podría tener efectos beneficiosos en términos de estancia hospitalaria y mortalidad.
El tratamiento actual de la sepsis se ha centrado en atenuar la magnitud de la respuesta del huésped. Sin embargo, pudiera ser más eficaz estimular la inmunidad del paciente para intentar erradicar de una manera más efectiva el patógeno causante de la infección. Se sabe que las citocinas antiinflamatorias son responsables de la regulación a la baja de la inmunidad celular y humoral, lo que provoca un período de relativa inmunodepresión denominado inmunoparálisis o síndrome de respuesta antiinflamatoria compensadora (compensatory anti-inflamatory response syndrome). Además, a medida que los neutrófilos son reclutados desde el espacio vascular al espacio intrapulmonar, el recuento de PMN circulantes puede disminuir; por lo que la liberación continuada de nuevos neutrófilos desde la médula ósea es crucial para el mantenimiento de una respuesta defensiva adecuada, máxime si se tiene en cuenta la reducida vida media circulante de estas células (4-8 h). Finalmente, se ha comprobado la existencia de defectos significativos en la inmunidad innata y adquirida en respuesta a la inflamación sistémica, lo que ha suscitado el desarrollo de otros abordajes menos tradicionales en el manejo de la sepsis. Todo esto ha promovido, en primer lugar, el empleo de agentes inmunomoduladores en el tratamiento de la neumonía. Así, el factor estimulante de las colonias granulocíticas (G-CSF) puede modificar las defensas del huésped incrementando el aflujo de leucocitos PMN al lugar de la infección, junto con una actividad antibacteriana específica32. El G-CSF es producido por múltiples células y su síntesis se inicia por diferentes estímulos proinflamatorios, como el LPS, que es un potente inductor de su expresión, o citocinas como el TNF-α, la IL-1β y la IL-17. Los efectos de señalización del G-CSF sobre los neutrófilos incluyen quimiotaxis, la expresión de moléculas de adhesión, la fagocitosis y el estallido respiratorio, aunque el efecto estimulador de esta citocina sobre la granulocitopoyesis es quizá el más importante durante la infección7. Como consecuencia de todo ello, el G-CSF se emplea habitualmente para reestablecer las concentraciones de PMN en pacientes neutropénicos. También se han realizado ensayos clínicos en pacientes con neumonía multilobular grave. En uno de ellos33 se aleatorizó a 480 pacientes con NAC multilobular para recibir G-CSF (n = 237) o placebo (n = 243). El tratamiento con G-CSF fue bien tolerado, aumentó el recuento leucocitario y mostró una tendencia, no estadísticamente significativa, hacia una menor mortalidad. En otro ensayo clínico multicéntrico más reciente34 los pacientes que recibieron 300 μg/día de G-CSF durante 5 días, o hasta que el recuento leucocitario alcanzó un mínimo de 75 x 109 células/l, no tuvieron una mortalidad menor que los que recibieron placebo. Sin embargo, sí hubo un efecto beneficioso, aunque no estadísticamente significativo, sobre la mortalidad cuando los pacientes recibían de manera simultánea G-CSF y una quinolona, en comparación con los pacientes que recibían placebo y esa misma clase de antibióticos (el 29 frente al 40%). No está del todo claro si el beneficio potencial del tratamiento con G-CSF es exclusivamente atribuible al incremento del número de neutrófilos en el lugar de la infección o si se debe también a un estímulo en la función de éstos. Además, el G-CSF aumenta la capacidad de los PMN para captar ciertos antimicrobianos, por lo que parece razonable conjeturar que ciertos antibióticos puedan ser mejor dirigidos hacia el tejido infectado si se administran en combinación con el G-CSF. En general, el empleo de G-CSF como tratamiento adyuvante en la neumonía bacteriana debería abordarse con cautela para evitar la descomposición del fino equilibrio en el que operan las defensas del huésped.
Igualmente se ha comprobado que la administración intrapulmonar de TNF-α y de IFN-α puede revertir la incapacidad de eliminar microorganismos inducida por la sepsis y restaurar la respuesta antimicrobiana del macrófago alveolar35,36. La implicación clínica de estos hallazgos en humanos todavía es desconocida. Se tiene alguna experiencia en la administración en aerosol de IFN-γ a pacientes con tuberculosis multirresistente, en quienes se han comprobado la tolerancia de la molécula y su efecto tanto en el incremento de peso como en la reducción de las lesiones cavitadas y en el tiempo de la negativización de los esputos37. Asimismo, se ha documentado un efecto beneficioso de la sobreexpresión compartimentada de IL-12 mediante genoterapia recombinante con adenovirus en un modelo murino de neumonía por Klebsiella38. Los efectos beneficiosos de la sobreexpresión de IL-12 están parcialmente mediados por la producción endógena de TNF-α y de IFN-α. En su conjunto, lo que indican estos trabajos es que en los procesos infecciosos graves la inmunoestimulación mediada por citocinas puede ser una estrategia mejor que el tratamiento antiinflamatorio en el manejo de pacientes seleccionados, y que su empleo sólo estaría limitado por la toxicidad relacionada con la dosis, especialmente cuando se administra por vía sistémica.
En los últimos años, el LPS bacteriano ha suscitado interés como molécula diana para el desarrollo de nuevos compuestos antimicrobianos, no sólo por su papel fundamental en la estabilidad de la membrana externa de los gramnegativos y, por tanto, en la viabilidad celular, sino también por su capacidad de iniciar una cascada de acontecimientos bioquímicos y celulares que provocan inflamación, disfunción orgánica y muerte. Los nuevos tratamientos anti-LPS incluyen aquellos que interrumpen su síntesis, los que previenen las interacciones entre LPS y las células efectoras del huésped, y los que se unen a LPS y neutralizan su actividad39. Entre los primeros se encuentran los agentes antilípido A o desacetilinas, que mediante la inhibición de una metaloenzima interfieren en la biosíntesis del lípido A, un componente esencial para el crecimiento de las bacterias gramnegativas. Además, estos agentes provocan un aumento de la permeabilidad de la membrana externa de la bacteria a pequeñas moléculas, lo que puede tener un efecto sinérgico con los antimicrobianos. Finalmente, otra de sus ventajas es que su actividad se limita exclusivamente a algunas clases de bacterias gramnegativas. Entre los agentes que bloquean la unión del LPS con su receptor se encuentran los análogos del lípido A, que carecen de la capacidad, o la tienen muy reducida, de activar estos receptores. Se ha documentado que el eritorán análogo de lípido A bloquea el efecto del LPS en un modelo humano de sepsis y que, en voluntarios sanos, inhibe la producción de TNF-α de forma dependiente de la dosis40. También se ha propuesto la utilización terapéutica de los TLR-4 solubles o la utilización de anticuerpos bloqueantes de la función de los TLR-4 (bloqueando la unión del LPS o interfiriendo con los correceptores MD-2 y CD14)41. Por último, se han desarrollado moléculas que se unen al LPS y neutralizan su actividad, como el factor recombinante LALF (factor Limulus anti-LPS), que es una proteína derivada del cangrejo cacerola americano que neutraliza LPS procedente de muy diferentes especies de gramnegativos debido a su afinidad por la mitad tóxica del lípido A42. La BPI, proteína ligadora que incrementa la permeabilidad, es una molécula producida fundamentalmente por los leucocitos, que actúa sobre la pared bacteriana incrementando su permeabilidad. Es, por consiguiente, bactericida para muchos patógenos gramnegativos. Hasta la fecha no se ha publicado ningún estudio que demuestre que la BPI recombinante sea útil en el tratamiento de la neumonía en humanos, aunque en un ensayo clínico para evaluar la eficacia de la BPI21 recombinante en pacientes con hemorragia traumática se documentó una menor proporción de neumonías y síndrome de distrés respiratorio agudo en los tratados en comparación con los controles43.
A pesar de lo esperanzadoras que parecen algunas de estas formas terapéuticas, la perspectiva más prometedora radicaría en saber cómo usar los agentes farmacológicos ya disponibles o desarrollados. El empleo potencial de marcadores genéticos para individualizar el tratamiento es particularmente pertinente para el uso del tratamiento inmunomodulador en la neumonía grave o en la indicación de vacunación en sujetos portadores de genotipos de riesgo. Ya se dispone de la tecnología para la genotipificación a tiempo real, y conocer qué variantes génicas son importantes para la toma de decisiones terapéuticas es el aspecto en el que se debe profundizar. Una vez que estas variantes se conozcan o se sospechen, deberán diseñarse ensayos clínicos con pacientes estratificados por su genotipo para confirmar el efecto beneficioso de un determinado agente terapéutico en un determinado genotipo. Sólo entonces podremos hacernos una idea real de la importancia de los estudios que hemos iniciado.
Trabajo financiado en parte por la Sociedad Española de Neumología y Cirugía Torácica (SEPAR) y por el Fondo de Investigaciones Sanitarias (PI04-1190 y PI 05-1196).
Correspondencia: Dr. F. Rodríguez de Castro.
Servicio de Neumología. Hospital Universitario de Gran Canaria Dr. Negrín.
Barranco de la Ballena, s/n. 35010 Las Palmas de Gran Canaria. Las Palmas. España.







