






En los últimos años se han producido importantes avances en el diagnóstico y tratamiento de la hipertensión pulmonar que han logrado una mejoría significativa en la supervivencia de esta enfermedad. Estas innovaciones se han recogido en guías de práctica clínica basadas en la evidencia elaboradas por las sociedades científicas. Sin embargo, no se incluyen en ellas, por falta de evidencia científica concluyente, algunos aspectos que inciden en la práctica asistencial. Conscientes de ello, la Sociedad Española de Neumología y Cirugía Torácica (SEPAR) y la Sociedad Española de Cardiología (SEC) han promovido la elaboración de un documento de consenso para definir en nuestro medio los estándares de calidad adecuados para el diagnóstico y tratamiento de la hipertensión pulmonar en sus diversas formas de presentación, así como la vía clínica y las directrices básicas de la organización asistencial del cuidado de estos pacientes, haciendo especial hincapié en los requisitos y funciones de las unidades de referencia. Para su redacción la SEPAR y la SEC designaron a un grupo de trabajo formado por expertos en los distintos aspectos de la enfermedad. Para la elaboración del documento se han utilizado las guías clínicas internacionales existentes, la revisión de la evidencia científica disponible y el debate en panel entre los expertos. El documento final, aprobado por todos los participantes, ha sido evaluado por revisores externos.
Substantial progress in the diagnosis and treatment of patients with pulmonary hypertension in recent years has led to significant improvement in survival. Evidence-based clinical practice guidelines issued by scientific societies reflect these new developments. However, certain clinically relevant issues have not been covered in consensus guidelines because of the lack of conclusive scientific evidence. Therefore, the Spanish Society of Pulmonology and Thoracic Surgery (SEPAR) and the Spanish Society of Cardiology (SEC) have promoted the present consensus statement in order to define national standards of care in the evaluation and management of pulmonary hypertension in its various forms, as well as to outline a clinical pathway and the basic principles for organizing health care in this clinical setting, with special emphasis on the requirements for and functions of specialized referral units. To prepare the statement, SEPAR and SEC formed a task force composed of national experts in various aspects of pulmonary hypertension. The resulting consensus is based on international clinical guidelines, a review of available scientific evidence, and panel discussion among the task force members. The final statement, approved by all participants, underwent external review.
En los últimos años se han producido importantes avances en la atención clínica de la hipertensión pulmonar, especialmente en el tratamiento, que han dado lugar a una mejoría significativa en las expectativas de supervivencia de los pacientes con las formas más graves de la enfermedad. Estas innovaciones se han reflejado en guías de práctica clínica basadas en la evidencia elaboradas por sociedades científicas internacionales1–4, que constituyen la referencia actual para el diagnóstico y tratamiento de la enfermedad. sin embargo, en dichas guías no se contemplan, por falta de evidencia científica concluyente, algunos aspectos que inciden en la práctica asistencial. por otro lado, para el correcto diagnóstico y tratamiento de la hipertensión pulmonar, especialmente en sus formas más graves, se requiere de técnicas y personal especializado con experiencia sólida en la enfermedad. A fin de atender a estas necesidades es conveniente que en nuestro país haya una estructura asistencial adecuada a los estándares de calidad exigibles para el manejo clínico de pacientes con hipertensión pulmonar.
Conscientes de ello, la sociedad Española de Neumología y Cirugía Torácica (SEPAR) y la Sociedad Española de cardiología (sEc) han promovido la elaboración de un documento que estableciera en nuestro medio los estándares de calidad adecuados para el diagnóstico y tratamiento de la hipertensión pulmonar en sus diversas formas de presentación, así como la vía clínica que deben seguir los pacientes, y que, por otro lado, aportara criterios razonados para la organización de la asistencia de los pacientes, de acuerdo con la especialización necesaria para proporcionar unos cuidados adecuados, teniendo en cuenta la prevalencia de la enfermedad.
Para la elaboración de dicho documento, la SEPAR y la SEC designaron sendos coordinadores. Éstos, a su vez, solicitaron la incorporación de especialistas en distintos aspectos de la enfermedad para formar un grupo de trabajo con composición paritaria entre ambas sociedades. Para la elaboración del documento de consenso se han combinado las indicaciones de las guías clínicas internacionales existentes, especialmente la de la European Society of Cardiology1, la revisión de la evidencia científica disponible y el debate en panel entre los expertos. El documento fue redactado por los coordinadores y revisado por todos los miembros del grupo de trabajo en repetidas ocasiones hasta su redacción final. El documento final ha sido evaluado por 2 revisores externos y aprobado por los órganos científicos y directivos de la SEPAR y la SEC.
2Definición y clasificaciónLa hipertensión pulmonar (HP) se define por el incremento anómalo de la presión en la arteria pulmonar. Por consenso se considera que existe HP cuando la presión media en la arteria pulmonar (PAPm) es igual o superior a 25 mmHg en reposo, o a 30 mmHg durante la realización de ejercicio.
La HP puede presentarse en distintos procesos clínicos o enfermedades que, de acuerdo con la clasificación actual1, se agrupan en 5 clases o categorías: I, arterial (hipertensión arterial pulmonar); II, asociada a enfermedad cardíaca izquierda; iii, asociada a enfermedad respiratoria y/o a hipoxemia; iV, secundaria a enfermedad tromboembólica, y V, grupo misceláneo. En la tabla I se indican las enfermedades y procesos clínicos incluidos en cada categoría. Esta clasificación está basada fundamentalmente en datos clínicos. Los procesos y enfermedades que se agrupan en las diferentes categorías comparten similitudes en los mecanismos fisiopatológicos, la presentación clínica y las opciones terapéuticas.
Clasificación de la hipertensión pulmonar
|
De entre las distintas clases de HP, las que tienen peor pronóstico, y por consiguiente requieren un proceso diagnóstico y un tratamiento más intensivos, son la hipertensión arterial pulmonar (HAP) y la HP tromboembólica crónica (HPTEC). En estas 2 categorías, a la definición hemodinámica de HP se añade la especificación de que la presión de oclusión de la arteria pulmonar (POAP) debe ser inferior a 16 mmHg y la resistencia vascular pulmonar superior a 3 mmHg·l-1·min-1 (unidades Wood) o 240 din·s·cm-5.
3Proceso diagnósticoEl diagnóstico de la HP requiere un proceso gradual dirigido a su identificación, clasificación y evaluación. En dicho proceso se distinguen 4 estadios: I, sospecha; II, detección; III, identificación de clase o categoría; y IV, evaluación y diagnóstico (tabla II).
Proceso diagnóstico de la hipertensión pulmonar
| Fase | Exploraciones |
| Sospecha | Síntomas, examen físico, radiografía de tórax, electrocardiograma |
| Detección | ETT |
| Identificación de clase y tipo |
|
| Evaluación y diagnóstico |
|
ETT: ecocardiograma transtorácico; HPTEC: hipertensión pulmonar tromboembólica crónica; TC: tomografía computarizada; VIH: virus de la inmunodeficiencia humana.
La sospecha de HP es eminentemente clínica y se fundamenta en los síntomas, la presencia de factores de riesgo, los hallazgos de la exploración física y los resultados de exámenes simples como la radiografía de tórax y el electrocardiograma.
La herramienta fundamental para la detección de la HP es la ecocardiografía transtorácica (ETT), por lo que debe practicarse siempre que se sospeche HP. La ETT permite estimar la presión sistólica arterial pulmonar (PSAP) a partir de la velocidad de regurgitación de la tricúspide y proporciona información sobre posibles causas cardíacas de la HP, así como de sus consecuencias sobre la morfología y función del ventrículo derecho. Se considera que puede haber HP cuando la velocidad de regurgitación tricuspídea es mayor de 2,8 m·s-1, valor que equivale, aproximadamente, a una PSAP superior a 36 mmHg. Debe tenerse en cuenta, sin embargo, que dicho valor aumenta de forma fisiológica con la edad5. La ETT permite detectar una posible HP, pero en ningún caso establecer su diagnóstico, ya que éste sólo es posible mediante el estudio hemodinámico pulmonar con cateterismo cardíaco derecho. Ante una sospecha clínica evidente de HP sin PSAP elevada en la ETT es aconsejable, dada la posibilidad de un falso negativo, realizar un estudio hemodinámico pulmonar para descartarla. Los parámetros que deben medirse en la ETT están convenientemente estandarizados y se resumen en la tabla III.
Parámetros ecocardiográficos que deben registrarse en el estudio diagnóstico de los pacientes con hipertensión pulmonar
|
AD: aurícula derecha; PSAP: presión sistólica arterial pulmonar; VD: ventrículo derecho; VI: ventrículo izquierdo.
Una vez detectada la posible HP, se inicia la fase de identificación de la clase o categoría de acuerdo con su clasificación (tabla I). Para ello se efectuarán los exámenes indicados en la tabla II.
Tras la detección ecocardiográfica de HP y la realización de los exámenes señalados, los pacientes con HAP, HPTEC y aquéllos con neumopatía con valores de PSAP desproporcionados para la gravedad del proceso subyacente deben ser remitidos a una unidad de referencia en HP a fin de completar el proceso diagnóstico.
La evaluación diagnóstica de la HP se completa con: a) el estudio hemodinámico pulmonar; b) la evaluación de la capacidad de ejercicio, y c) exámenes específicos según el tipo de HP. El estudio hemodinámico pulmonar mediante cateterismo cardíaco derecho es el examen de referencia para el diagnóstico de HP. Los resultados obtenidos permiten evaluar la gravedad de la HP y contribuyen a establecer el pronóstico. Su realización está indicada en el diagnóstico de todos los pacientes con HAP o HPTEC (categorías I y IV, respectivamente). En otras circunstancias su realización está justificada si de sus resultados se derivará un cambio en la situación clínica del paciente o del tratamiento. En pacientes con PSAP menor de 50 mmHg estimada por ETT se deberá individualizar la decisión en función de la edad, el fundamento de la sospecha clínica y la comorbilidad.
El estudio hemodinámico diagnóstico debe acompañarse de la valoración de la respuesta vasodilatadora aguda, para lo cual se empleará uno de los agentes recomendados (epoprostenol intravenoso, óxido nítrico inhalado o adenosina intravenosa). El resultado de la prueba vasodilatadora tiene importantes implicaciones en el tratamiento y el pronóstico. Se considera que es positiva cuando se produce un descenso de la PAPm de como mínimo 10 mmHg, con un valor de PAPm final igual o inferior a 40 mmHg, sin que se produzca descenso del gasto cardíaco.
Tanto el estudio hemodinámico pulmonar como la prueba vasodilatadora pulmonar deben ser realizados por personal experimentado, en unidades dotadas con el equipamiento adecuado.
La evaluación del paciente con HP diagnosticada se completa con la valoración de la gravedad del proceso. Para ello se tienen en cuenta: a) la clase funcional, de acuerdo con la escala de la New York Heart Association (NYHA) modificada por la Organización Mundial de la Salud (OMS)1; b) la tolerancia al ejercicio; c) los resultados de los estudios ecocardiográfico y hemodinámico, y d) la analítica sanguínea. La tolerancia al ejercicio se evalúa habitualmente mediante la prueba de marcha de 6 min, que debe realizarse siguiendo las recomendaciones establecidas6. Se aconseja interpretar los resultados obtenidos comparándolos con valores de referencia en los que se tengan en cuenta el sexo, la edad y la estatura del paciente7. En pacientes en clases funcionales I-II el resultado de la prueba de marcha de 6 min puede ser normal. En esta situación la prueba de esfuerzo cardiopulmonar puede aportar información más precisa acerca de la tolerancia al ejercicio y los factores que la limitan.
3.1Situaciones especiales- –
En la HAP familiar, si hay antecedentes familiares de HP es conveniente estudiar la posible presencia de mutaciones del gen BMPR2 en el paciente diagnosticado (caso índice). Si se identifican mutaciones en el caso índice, se aconseja ampliar el estudio al resto de familiares con el objeto de detectar precozmente HP en portadores asintomáticos.
- –
El diagnóstico de HPTEC se establece por la presencia de fenómenos trombóticos pulmonares e HP tras más de 3 meses de tratamiento anticoagulante correcto. En estos pacientes es preciso determinar si las lesiones trombóticas tienen una localización proximal o distal. Para ello la prueba radiológica de referencia es la angiografía pulmonar selectiva. La angiografía por tomografía computarizada helicoidal efectuada con equipos de última generación también proporciona imágenes de gran fidelidad, aunque actualmente se considera complementaria a la angiografía convencional y no la sustituye. La valoración de los pacientes con HPTEC debe efectuarse en centros especializados en dicha enfermedad, preferiblemente con experiencia en tromboendarterectomía pulmonar.
- –
En los pacientes con HP asociada a cardiopatía izquierda la elevación de la PSAP en el ecocardiograma no constituye por sí misma una indicación de estudio hemodinámico. El cateterismo derecho se realizará cuando haya dudas diagnósticas sobre si la HP detectada en el ecocardiograma es HAP (HP precapilar) o HP asociada con enfermedad cardíaca izquierda (HP poscapilar), generalmente pacientes con función sistólica normal y únicamente disfunción diastólica del ventrículo izquierdo.
- –
En los pacientes con HP asociada a enfermedad respiratoria tampoco es necesario el estudio hemodinámico pulmonar, excepto en los casos en que el valor de la PSAP estimado por ETT sea desproporcionadamente elevado (> 55 mmHg) en relación con la gravedad de la enfermedad respiratoria y se considere que la HP puede constituir un proceso concomitante, potencialmente tributario de tratamiento específico.
- –
No es preciso efectuar estudio polisomnográfico a todos los pacientes con HP como cribado de un posible síndrome de apneas del sueño, salvo que haya sospecha clínica.
Las medidas generales contemplan estrategias destinadas a disminuir el impacto nocivo de algunas circunstancias y agentes externos en los pacientes con HP.
- –
El ejercicio físico puede aumentar la PAPm, por lo que debe evitarse aquel que produzca síntomas graves (síncope o presíncope). El ejercicio aeróbico suave y progresivo, con una frecuencia de 4–5 días a la semana, es recomendable.
- –
En los vuelos comerciales se aconseja el uso de oxígeno suplementario si se realiza un viaje prolongado (> 2 h) o si hay insuficiencia respiratoria.
- –
La gestación produce cambios hormonales y hemodinámicos que suelen ser muy mal tolerados. La mortalidad materna en esta situación es elevada (30-50%), especialmente en el posparto inmediato. Por ello es imprescindible la utilización de métodos anticonceptivos. Se desaconsejan los anticonceptivos hormonales combinados por su posible efecto protrombótico, siendo de elección los métodos de barrera y los anticonceptivos hormonales sin estrógenos. La esterilización quirúrgica en mujeres y la implantación de dispositivos intrauterinos requieren seguimiento y anestesia especializada por la posibilidad de complicaciones (reacciones vasovagales) potencialmente fatales. En caso de embarazo, se recomienda su interrupción durante el primer trimestre.
- –
Oxigenoterapia continua domiciliaria. Debe prescribirse cuando haya insuficiencia respiratoria, tratando de mantener una saturación arterial de oxígeno superior al 90%.
- –
Diuréticos. Están indicados para la reducción de los signos y síntomas de insuficiencia cardíaca derecha. La espironolactona está especialmente recomendada.
- –
Digital. Se utilizará en casos con insuficiencia ventricular derecha clínicamente evidente o fibrilación auricular.
- –
Anticoagulantes orales. Se recomienda la anticoagulación en pacientes con HAP idiopática y formas asociadas, a excepción de la hipertensión portopulmonar con varices esofágicas. En el síndrome de Eisenmenger la anticoagulación es objeto de debate. Se recomienda mantener un cociente internacional normalizado (INR) entre 1,5 y 2,5.
- –
Vasodilatadores antagonistas del calcio. La administración crónica de dosis altas de antagonistas del calcio prolonga la supervivencia de los pacientes que presentan respuesta significativa en la prueba vasodilatadora aguda. Los fármacos más utilizados son el diltiazem y el nifedipino. Las dosis a las que han demostrado ser eficaces son
- –
relativamente elevadas: 240–720 mg/día de diltiazem y 120–240 mg/día de nifedipino. La eficacia de los antagonistas del calcio debe evaluarse a los 3–6 meses de su inicio. Se considerará que el tratamiento es eficaz si la clase funcional es I o II y la PSAP cercana a los valores normales. Si no se consiguen estos objetivos, está indicado iniciar tratamiento con fármacos específicos.
La utilidad del tratamiento a largo plazo con antagonistas del calcio en pacientes con HAP asociada a colagenosis o infección por el virus de la inmunodeficiencia humana (VIH) que han respondido significativamente en la prueba vasodilatadora es menos clara. No hay indicación de tratamiento con antagonistas del calcio en el síndrome de Eisenmenger y la hipertensión portopulmonar.
4.1.2.2Fármacos específicos (tabla IV)4.1.2.2.1Prostanoides- –
Epoprostenol. Es el fármaco del que se dispone de mayor experiencia clínica, el que ha demostrado más claramente su eficacia en pacientes con HAP en clase funcional IV y del que hay evidencias más sólidas en cuanto al incremento de la supervivencia. Se administra por vía intravenosa continua a través de un catéter central permanente, con las complicaciones que ello puede conllevar. La interrupción de la administración puede producir efecto de rebote y graves complicaciones, y por tanto debe reinstaurarse en el período más breve posible.
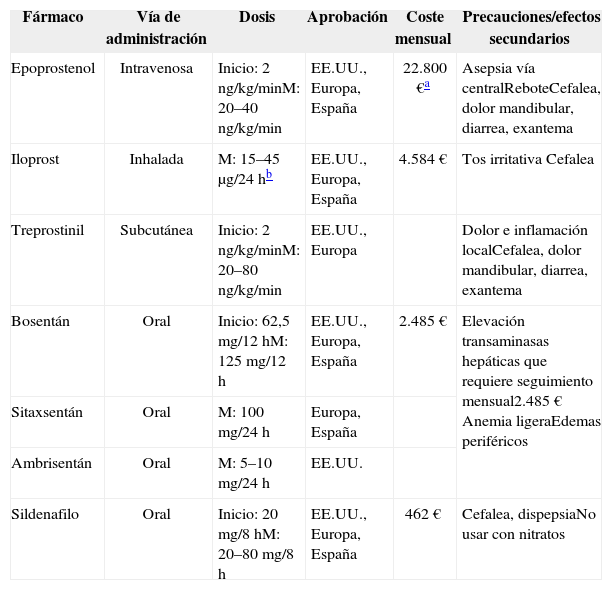
Tabla IV.Fármacos específicos para el tratamiento de la hipertensión pulmonar
Fármaco Vía de administración Dosis Aprobación Coste mensual Precauciones/efectos secundarios Epoprostenol Intravenosa Inicio: 2 ng/kg/minM: 20–40 ng/kg/min EE.UU., Europa, España 22.800 €a Asepsia vía centralReboteCefalea, dolor mandibular, diarrea, exantema Iloprost Inhalada M: 15–45 μg/24 hb EE.UU., Europa, España 4.584 € Tos irritativa Cefalea Treprostinil Subcutánea Inicio: 2 ng/kg/minM: 20–80 ng/kg/min EE.UU., Europa Dolor e inflamación localCefalea, dolor mandibular, diarrea, exantema Bosentán Oral Inicio: 62,5 mg/12 hM: 125 mg/12 h EE.UU., Europa, España 2.485 € Elevación transaminasas hepáticas que requiere seguimiento mensual2.485 € Anemia ligeraEdemas periféricos Sitaxsentán Oral M: 100 mg/24 h Europa, España Ambrisentán Oral M: 5–10 mg/24 h EE.UU. Sildenafilo Oral Inicio: 20 mg/8 hM: 20–80 mg/8 h EE.UU., Europa, España 462 € Cefalea, dispepsiaNo usar con nitratos M: mantenimiento.
- –
Iloprost. Es un análogo de la prostaciclina con una vida media sérica de 20–25 min, que se administra por vía inhalada. Se requieren de 6 a 9 sesiones de inhalación diarias. También puede administrarse por vía intravenosa, aunque la experiencia clínica es escasa.
- –
Treprostinil. Es el análogo de prostaciclina con vida media más larga (2–3 h), lo cual permite su administración por vía subcutánea. El principal efecto secundario es el dolor y la inflamación local en el punto de infusión, que requiere medidas terapéuticas específicas y puede obligar al cambio del tratamiento. Se puede administrar también por vía intravenosa, aunque la experiencia es escasa. Actualmente se están realizando ensayos clínicos para validar la eficacia de su administración por vía inhalada y oral.
- –
Bosentán. Es un antagonista dual de los receptores A y B de la endotelina-1, que se administra por vía oral. Puede producir toxicidad hepática (incrementos superiores al triple del valor normal de transaminasas) reversible en el 8% de los pacientes, lo que obliga al seguimiento mensual del perfil hepático. Tiene efectos teratógenos, por lo que está contraindicado en el embarazo. Interacciona con los anticonceptivos hormonales orales, disminuyendo su eficacia, y con la glibenclamida, incrementando el riesgo de toxicidad hepática.
- –
Sitaxsentán. Es un antagonista selectivo del receptor A de la endotelina-1, recientemente autorizado, por lo que la experiencia en su uso es todavía escasa.
- –
Ambrisentán. Es un antagonista selectivo del receptor A de la endotelina-1. Se han completado recientemente los ensayos clínicos controlados y la Food and Drug Administration estadounidense ha aprobado su uso (junio de 2007).
- –
Sildenafilo. Es un inhibidor de la fosfodiesterasa-5 que se administra por vía oral. La dosis recomendada es de 20 mg/8 h, aunque se ha empleado en dosis hasta 4 veces superiores. Interacciona con algunos fármacos antirretrovirales y exige ajuste de dosis. Nunca puede utilizarse conjuntamente con nitratos por riesgo de hipotensión grave.
- –
Tadalafilo. Es un inhibidor más potente de la fosfodiesterasa-5. Actualmente se está completando un ensayo clínico controlado de eficacia en la HAP.
Las indicaciones y recomendaciones de los fármacos específicos para el tratamiento de la HP en monoterapia se han dividido en 4 categorías, teniendo en cuenta el grado de evidencia disponible, la autorización por agencias reguladoras8, las recomendaciones de las guías clínicas1–4 y la información científica existente:
- 1.
Indicación establecida en guías clínicas actuales y autorizada por agencias reguladoras de medicamentos.
- 2.
Aceptable: indicaciones aceptadas por expertos y que forman parte de la práctica clínica habitual, no comprobadas todavía mediante ensayos clínicos controlados, pero con alguna evidencia de eficacia en estudios no controlados.
- 3.
Experimental: indicaciones no reconocidas ni validadas en la comunidad científica, por lo que deben considerarse experimentales. Se recomienda su utilización únicamente por personal experto en el tratamiento de la HP, preferiblemente en el contexto de ensayos clínicos o registros que permitan analizar los resultados obtenidos.
- 4.
No recomendado: indicaciones no reconocidas ni validadas por la comunidad científica que se consideran no apropiadas.
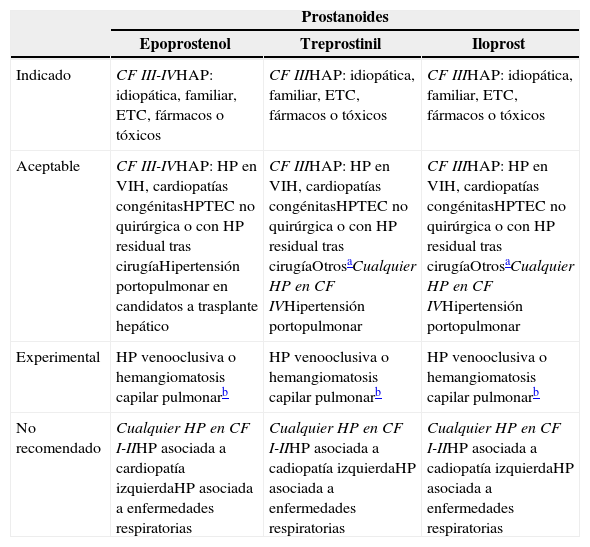
Fármacos específicos para el tratamiento de la hipertensión pulmonar (HP)
| Prostanoides | |||
| Epoprostenol | Treprostinil | Iloprost | |
| Indicado | CF III-IVHAP: idiopática, familiar, ETC, fármacos o tóxicos | CF IIIHAP: idiopática, familiar, ETC, fármacos o tóxicos | CF IIIHAP: idiopática, familiar, ETC, fármacos o tóxicos |
| Aceptable | CF III-IVHAP: HP en VIH, cardiopatías congénitasHPTEC no quirúrgica o con HP residual tras cirugíaHipertensión portopulmonar en candidatos a trasplante hepático | CF IIIHAP: HP en VIH, cardiopatías congénitasHPTEC no quirúrgica o con HP residual tras cirugíaOtrosaCualquier HP en CF IVHipertensión portopulmonar | CF IIIHAP: HP en VIH, cardiopatías congénitasHPTEC no quirúrgica o con HP residual tras cirugíaOtrosaCualquier HP en CF IVHipertensión portopulmonar |
| Experimental | HP venooclusiva o hemangiomatosis capilar pulmonarb | HP venooclusiva o hemangiomatosis capilar pulmonarb | HP venooclusiva o hemangiomatosis capilar pulmonarb |
| No recomendado | Cualquier HP en CF I-IIHP asociada a cardiopatía izquierdaHP asociada a enfermedades respiratorias | Cualquier HP en CF I-IIHP asociada a cadiopatía izquierdaHP asociada a enfermedades respiratorias | Cualquier HP en CF I-IIHP asociada a cadiopatía izquierdaHP asociada a enfermedades respiratorias |
| Antagonistas de los receptores de la endotelina | Inhibidores de la fosfodiesterasa-5 | ||
| Bosentán | Sitaxsentán/ambrisentánc | Sildenafilo | |
| Indicado | CF IIIHAP: idiopática, familiar, ETC, fármacos o tóxicos y cardiopatías congénitas | CF IIIHAP: idiopática, familiar, ETC, fármacos o tóxicos | CF IIIHAP: idiopática, familiar, ETC, fármacos o tóxicos |
| Aceptable | CF IIHAP: idiopática, familiar, ETCCF IIIHAP: HP en VIHHPTEC no quirúrgica o con HP residual tras cirugíaOtrosaCualquier HP en CF IV | CF IIHAP: idiopática, familiar, ETCCF IIIHAP: HP en VIHHPTEC no quirúrgica o con HP residual tras cirugíaOtrosaCualquier HP en CF IV | |
| Experimental | HP venooclusiva o hemangiomatosis capilar pulmonarbHipertensión portopulmonar Child A o BHP desproporcionada (PAPm > 40 mmHg) en fibrosis pulmonarHP secundaria a cardiopatía izquierda, viabilidad trasplante cardíaco y perioperatorio de cirugía cardíaca | HP venooclusiva o hemangiomatosis capilar pulmonarbHipertensión portopulmonarHP desproporcionada (PAPm > 40 mmHg) en fibrosis pulmonar y EPOCHP secundaria a cardiopatía izquierda, viabilidad trasplante cardíaco y perioperatorio de cirugía cardíaca | |
| No recomendado | Cualquier HP en CF IHP asociada a cardiopatía izquierdaHP asociada a enfermedades respiratorias con PAPm < 40 mmHgHipertensión portopulmonar Child C | Cualquier HP en CF IHP asociada a cardiopatía izquierdaHP asociada a enfermedades respiratorias | Cualquier HP en CF IHP asociada a enfermedades respiratorias con PAPm < 40 mmHg |
CF: clase funcional de la New York Heart Association-Organización Mundial de la Salud; EPOC: enfermedad pulmonar obstructiva crónica; ETC: enfermedades del tejido conectivo; HAP: hipertensión arterial pulmonar; HPTEC: hipertensión pulmonar tromboembólica crónica; PAPm: presión media en la arteria pulmonar; VIH: virus de la inmunodeficiencia humana.
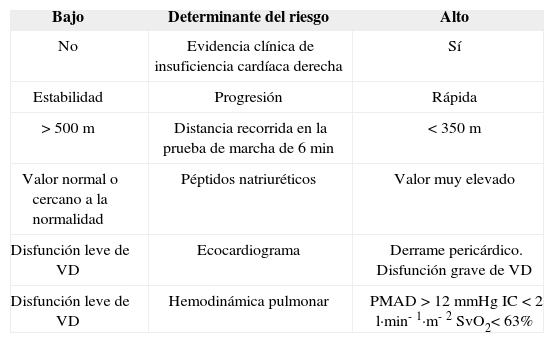
El tratamiento combinado con fármacos de distintas categorías puede estar indicado si con un régimen de monoterapia no se consigue situar al paciente en un perfil de bajo riesgo9 y aceptable control de la HP (tabla VI). El fracaso de la monoterapia es un criterio de derivación preferente a una unidad de referencia en HP.
Perfil de riesgo de evolución desfavorable en el tratamiento de la hipertensión pulmonar
| Bajo | Determinante del riesgo | Alto |
| No | Evidencia clínica de insuficiencia cardíaca derecha | Sí |
| Estabilidad | Progresión | Rápida |
| > 500 m | Distancia recorrida en la prueba de marcha de 6 min | < 350 m |
| Valor normal o cercano a la normalidad | Péptidos natriuréticos | Valor muy elevado |
| Disfunción leve de VD | Ecocardiograma | Derrame pericárdico. Disfunción grave de VD |
| Disfunción leve de VD | Hemodinámica pulmonar | PMAD > 12 mmHg IC < 2 l·min-1·m-2 SvO2< 63% |
IC: índice cardíaco; PMAD: presión media de aurícula derecha; SvO2: saturación de oxígeno en sangre venosa mezclada (arteria pulmonar); VD: ventrículo derecho.
No se dispone en este momento de información suficiente sobre la combinación más eficaz, su dosificación y los efectos secundarios que pueden producirse. Por ello se recomienda extremar la vigilancia y realizarlo sólo en las unidades de referencia en HP. En la actualidad hay varios ensayos clínicos en marcha para valorar la eficacia y seguridad de diferentes combinaciones de tratamientos.
4.1.3Tratamiento no farmacológico4.1.3.1Septostomía auricularConsiste en crear un cortocircuito derecha-izquierda a través de la fosa oval. Es un procedimiento paliativo que en general se utiliza como puente al trasplante pulmonar. La septostomía descomprime el ventrículo derecho e incrementa la precarga izquierda, mejorando el gasto cardíaco y el transporte tisular de oxígeno, a pesar del descenso de la PaO2. Su eficacia sólo se ha evaluado en series con escaso número de pacientes. La mortalidad inmediata del procedimiento oscila entre el 5 y el 13%. La septostomía auricular se debe realizar siempre en centros con experiencia.
Sus indicaciones son:
- 1.
Pacientes con HAP grave en clases III-IV, con síncope recurrente o insuficiencia cardíaca derecha, refractarios a tratamiento médico.
- 2.
Pacientes considerados para trasplante pulmonar, como puente o como tratamiento paliativo si no hay ninguna opción alternativa.
Las contraindicaciones, por la alta mortalidad asociada, son: situación de muerte inminente, saturación arterial inferior al 90% y hemoglobina menor de 12 g/dl.
4.1.4Trasplante pulmonar y cardiopulmonarEn los pacientes con enfermedad moderada o grave que no responden al tratamiento médico, el trasplante puede lograr la normalización de la hemodinámica pulmonar, con mejoría significativa de la clínica, la calidad de vida y la supervivencia. Estos beneficios se han demostrado en estudios no controlados de series de pacientes. La complejidad del tratamiento, el riesgo de mortalidad del procedimiento y la limitación de los resultados como consecuencia del rechazo crónico hacen que el trasplante se contemple como la última opción en el algoritmo terapéutico de la HP.
La elección del tipo de procedimiento (unipulmonar, bipulmonar o cardiopulmonar) depende de la enfermedad subyacente y de la situación hemodinámica. El procedimiento preferido es el trasplante bipulmonar, cuya supervivencia actuarial a los 5 años es del 50%. El tras plante cardiopulmonar está indicado cuando hay afectación cardíaca grave que desaconseja el trasplante pulmonar aislado (tabla VII).
Trasplante pulmonar y cardiopulmonar en la hipertensión pulmonar
|
La ventana temporal para la indicación de trasplante es estrecha y difícil de determinar. Se debe remitir a los pacientes para valoración a una unidad de trasplante cuando, cumpliendo los criterios generales para éste10, la enfermedad sea rápidamente progresiva o el perfil de evolución sea desfavorable (tabla VI) y se considere que podrá llegar a plantearse esta opción terapéutica. En líneas generales, en la HAP esta situación se plantea cuando, por la evolución clínica del paciente, se requiere la administración de epoprostenol intravenoso y la respuesta clínica a éste no es favorable; es decir, si a los 3 meses de su instauración la clase funcional es III-IV y la distancia recorrida en 6 min menor de 380 m11. En la enfermedad venooclusiva pulmonar y la hemangiomatosis capilar pulmonar el trasplante pulmonar representa la primera opción terapéutica al no disponerse de tratamiento médico de eficacia demostrada.
4.1.5Situaciones especialesHay formas de HAP y circunstancias especiales que requieren un manejo específico y son subsidiarias de recomendaciones diferenciadas:
El síndrome de Eisenmenger forma parte del grupo de cardiopatías congénitas cianóticas y, como tal, su manejo debe reservarse a facultativos expertos en dicho campo. Se debe contemplar el tratamiento de la vasculopatía
- 1.
Cardiopatías congénitas. De entre las situaciones en que la HP se asocia a las cardiopatías congénitas nos referiremos exclusivamente al síndrome de Eisenmenger, que se caracteriza por resistencia vascular pulmonar elevada con cortocircuito bidireccional o invertido. Hay guías clínicas acerca de esta entidad que deberán seguirse12. Los pacientes con síndrome de Eisenmenger tienen mejor pronóstico vital que los que presentan otras formas de HP, aunque pueden experimentar limitación funcional grave de forma prolongada.
El síndrome de Eisenmenger forma parte del grupo de cardiopatías congénitas cianóticas y, como tal, su manejo debe reservarse a facultativos expertos en dicho campo. Se debe contemplar el tratamiento de la vasculopatía arterial pulmonar y de las complicaciones propias de la cardiopatía subyacente, con los siguientes principios generales:
- –
Anticoagulación: se aconseja en pacientes con arritmias auriculares, trombosis de las arterias pulmonares o antecedentes de embolia. Debe ajustarse el INR a la poliglobulia, con un rango que oscile entre 1,5 y 2,5. Se desaconseja si hay antecedentes de hemoptisis significativa.
- –
Oxigenoterapia: no se ha descrito efecto beneficioso.
- –
Sangrías: sólo están indicadas si el hematocrito es mayor del 65% y hay síntomas de hiperviscosidad, o bien antes de la cirugía para la corrección de la coagulopatía asociada.
- –
Los fármacos específicos para el tratamiento de la HP se han utilizado con éxito en pacientes con síndrome de Eisenmenger. Sin embargo, la experiencia es escasa y los estudios publicados incluyen a un número reducido de pacientes, con tiempos de seguimiento cortos. Únicamente se ha realizado un ensayo clínico, con buenos resultados, con bosentán, durante un período de seguimiento breve.
- –
- 2.
Hipertensión portopulmonar. Es la asociación de HP e hipertensión portal, con o sin enfermedad hepática. Hemodinámicamente se define por una PAPm mayor de 25 mmHg, POAP menor de 15 mmHg y resistencia vascular pulmonar por encima de 240 din·s·cm-5. Los pacientes con hipertensión portopulmonar son los que presentan mayor mortalidad, que puede deberse tanto a causas hepáticas como ser consecuencia de la HP.
La presencia de HP condiciona mayor morbimortalidad en el trasplante hepático y lo contraindica si la PAPm es mayor de 35 mmHg13. En estos pacientes se retirará el tratamiento bloqueador beta y, si hay varices esofágicas, se efectuará ligadura con bandas de éstas para prevenir la hemorragia. No deben administrarse anticoagulantes.
No se han efectuado ensayos clínicos controlados. La única información disponible procede de series con escaso número de pacientes y períodos de seguimiento cortos, en las que se han empleado epoprostenol, bosentán o sildenafilo. Estos fármacos deben administrarlos personas con experiencia tanto en la HP como en la hemodinámica hepática, en unidades especializadas. El bosentán entraña el riesgo añadido de su potencial hepatotoxicidad y su empleo quedará restringido a pacientes con función hepática aceptable.
4.2Hipertensión pulmonar con enfermedad cardíaca izquierdaEs la causa más frecuente de HP, dado que está presente en el 30-40% de los pacientes con cardiopatía izquierda. La HP puede ser pasiva —POAP > 16 mmHg, gradiente transpulmonar (PAPm-POAP) < 12 mmHg— o reactiva —POAP > 16 mmHg, gradiente transpulmonar > 12 mmHg—. Ésta, a su vez, tiene 2 componentes:
- a)
reactivo y reversible con fármacos vasodilatadores, y
- b)
fijo, fruto de la remodelación vascular.
El tratamiento actual de la HP en las cardiopatías izquierdas es el propio de la enfermedad subyacente, dado que no se ha encontrado ningún fármaco específico, seguro y eficaz en el tratamiento crónico de la HP pasiva o reactiva de los pacientes con insuficiencia cardíaca. El tratamiento farmacológico de la HP queda restringido al ámbito perioperatorio, bien en la valoración de la reversibilidad de la HP previa a la cirugía o al trasplante cardíacos, o en el manejo de la HP reversible en el postoperatorio. Los fármacos utilizados para este fin son óxido nítrico, sildenafilo, prostanoides e inhibidores de la endotelina; de ellos, el óxido nítrico es el más usado en el postoperatorio de la cirugía cardíaca.
Se ha evaluado la utilidad de los inhibidores de la endotelina y de epoprostenol como forma de tratamiento crónico en ensayos clínicos controlados con pacientes que presentaban insuficiencia cardíaca, sin que se haya objetivado su eficacia.
4.3Hipertensión pulmonar asociada a enfermedad respiratoria o hipoxemiaEs también causa frecuente de HP, aunque la gravedad suele ser leve-moderada.
- –
Enfermedad pulmonar obstructiva crónica (EPOC). El tratamiento actualmente aceptado de la HP en la EPOC es el propio de la enfermedad de base y la oxigenoterapia continua domiciliaria. Se ha identificado a un subgrupo reducido de pacientes que presentan HP grave (PAPm > 40 mmHg), en quienes el valor de PAPm no guarda relación con la gravedad de la obstrucción al flujo aéreo o la hipoxemia14,15. Es incierto que en estos casos la HP sea debida a la propia EPOC, y se ha planteado que podría constituir un proceso intercurrente. En la EPOC no está indicado el tratamiento de la HP con antagonistas del calcio. La información disponible sobre el empleo de fármacos específicos es anecdótica. Debe tenerse en cuenta que algunos de estos fármacos (sildenafilo, prostaciclina) inhiben la vasoconstricción pulmonar hipóxica y pueden empeorar el intercambio gaseoso en la EPOC.
- –
Neumopatías intersticiales. En la mayoría de los casos la HP también suele ser ligera, excepto cuando concurren fibrosis pulmonar e HP asociadas a una enfermedad del tejido conectivo. La información disponible sobre el empleo de fármacos específicos en la HP asociada a fibrosis pulmonar es escasa y sólo hay series abiertas de pocos pacientes seguidos durante períodos de tiempo cortos. Actualmente se están realizando ensayos clínicos controlados en este sentido.
Si el paciente presenta una enfermedad del tejido conectivo en la que coexistan HP grave y fibrosis pulmonar moderada, se recomienda efectuar el tratamiento protocolizado de la HAP.
- –
Síndrome de apneas-hipopneas durante el sueño. Su importancia como causa de HP no está bien establecida. En los pacientes con síndrome de apneas-hipopneas durante el sueño e HP demostrada, el tratamiento con presión positiva continua de la vía aérea mejora sustancialmente la HP. Si no es así, debería considerarse que haya HAP concurrente.
La incidencia de la HPTEC tras una tromboembolia pulmonar es del 3,8% a los 2 años. La búsqueda de HPTEC debe realizarse sistemáticamente en todo paciente con HP, tenga o no antecedentes de tromboembolia pulmonar. El tratamiento anticoagulante es imprescindible (INR: 2,5-3,5).
El tratamiento específico de la HPTEC es la tromboendarterectomía pulmonar, siempre que se cumplan los siguientes requisitos:
- 1.
Clases funcionales III-IV de la NYHA-OMS.
- 2.
Resistencia vascular pulmonar mayor de 300 din·s·cm-5.
- 3.
Trombos organizados accesibles quirúrgicamente (arterias pulmonares principales, lobares o segmentarias).
- 4.
Ausencia de enfermedades asociadas graves.
La mortalidad del procedimiento oscila entre el 5 y el 24%, y está íntimamente relacionada con la curva de aprendizaje en la evaluación preoperatoria, la cirugía y los cuidados postoperatorios. Los centros con mayor volumen de actividad tienen menor mortalidad. La valoración de la indicación quirúrgica en un paciente con HPTEC debe realizarse en centros subespecializados en tromboendarterectomía pulmonar.
Los pacientes con HPTEC no tributarios de tratamiento quirúrgico son candidatos a tratamientos específicos (prostanoides, bosentán y sildenafilo). Se han publicado experiencias favorables con un reducido número de pacientes y períodos cortos de seguimiento con estos fármacos. Sin embargo, la mejoría hemodinámica obtenida ha sido claramente inferior a la conseguida con la tromboendarterectomía pulmonar. El tratamiento médico de estos pacientes debe realizarse en las unidades de referencia en HP.
5Seguimiento clínicoLos pacientes con HP como diagnóstico principal (HAP, HPTEC) deben ser controlados de forma estrecha con los siguientes objetivos: a) valorar la respuesta al tratamiento; b) prevenir las complicaciones; c) detectar precozmente el empeoramiento clínico, y d) modificar la pauta terapéutica de acuerdo con la evolución clínica.
Se ha comprobado que con algunos fármacos la respuesta a los 3 meses de iniciar el tratamiento es predictiva de la evolución a más largo plazo. Por este motivo se recomienda que a los 3 meses de instaurarlo o modificarlo se evalúen la situación clínica del paciente (clase funcional) y la tolerancia al ejercicio (prueba de marcha de 6 min). Alcanzar una clase funcional I o II, recorrer más de 380 m en la prueba de marcha de 6 min, un consumo pico de oxígeno mayor de 10,4 ml/kg/min y una cifra de presión arterial sistólica en el umbral láctico por encima de 120 mmHg en la prueba de esfuerzo cardiopulmonar son indicadores de pronóstico favorable11,16,17. En general, se recomienda mantener al paciente dentro del perfil de bajo riesgo de evolución desfavorable (tabla VI) durante el seguimiento. Actualmente no se dispone de información prospectiva que permita delimitar de forma precisa los factores pronósticos de respuesta al tratamiento.
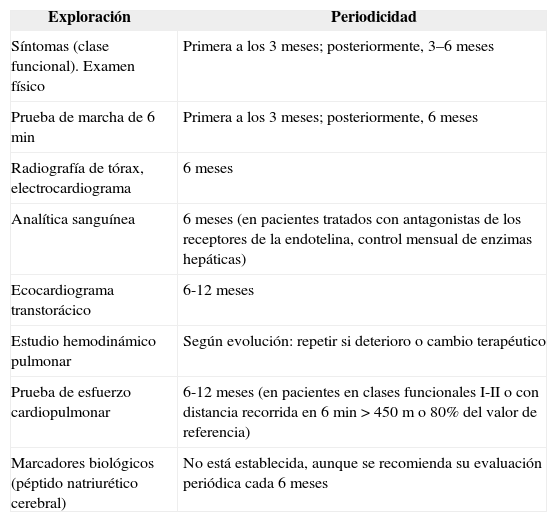
La periodicidad de las visitas de seguimiento viene dictada por el tipo de HP, la situación clínica del paciente, el régimen terapéutico instaurado y la respuesta a éste. En la tabla VIII se presentan recomendaciones orientativas al respecto.
Seguimiento clínico
| Exploración | Periodicidad |
| Síntomas (clase funcional). Examen físico | Primera a los 3 meses; posteriormente, 3–6 meses |
| Prueba de marcha de 6 min | Primera a los 3 meses; posteriormente, 6 meses |
| Radiografía de tórax, electrocardiograma | 6 meses |
| Analítica sanguínea | 6 meses (en pacientes tratados con antagonistas de los receptores de la endotelina, control mensual de enzimas hepáticas) |
| Ecocardiograma transtorácico | 6-12 meses |
| Estudio hemodinámico pulmonar | Según evolución: repetir si deterioro o cambio terapéutico |
| Prueba de esfuerzo cardiopulmonar | 6-12 meses (en pacientes en clases funcionales I-II o con distancia recorrida en 6 min > 450 m o 80% del valor de referencia) |
| Marcadores biológicos (péptido natriurético cerebral) | No está establecida, aunque se recomienda su evaluación periódica cada 6 meses |
La ETT proporciona información sobre la morfología y función del ventrículo derecho que tiene significación pronóstica y es útil para el seguimiento de los pacientes. El valor de la PSAP guarda poca relación con el pronóstico. Se recomienda repetir la ETT cada 6–12 meses.
El estudio hemodinámico pulmonar efectuado a los 3 meses de instaurar un tratamiento específico tiene significación pronóstica, especialmente el cambio en el gasto cardíaco. Sin embargo, dada la invasividad del procedimiento, no se recomienda que se repita dicho estudio de forma sistemática, sino atendiendo a la evolución clínica del paciente, especialmente si se deteriora o se considera la posibilidad de modificar la pauta terapéutica.
6Organización asistencial: unidades de referencia en hipertensión pulmonarLa HP, sobre todo las formas HAP y HPTEC, precisa de cuidados de elevado grado de especialización, lo cual requiere concentrar los casos a tratar en un número reducido de unidades de referencia. La necesidad de derivar los casos en que se sospeche HAP o HPTEC a unidades de referencia especializadas en esta enfermedad viene recogida en todas las guías clínicas actuales1–3. La pertinencia de crear unidades de referencia en HP en nuestro país está justificada por los siguientes motivos:
- 1.
Es una enfermedad poco prevalente. La HAP tiene una prevalencia de 15 casos por 1.000.000 de habitantes18, con lo que cumple los criterios de enfermedad rara. Se estima que en España pueden presentar HAP entre 600 y 800 personas.
- 2.
La HAP y la HPTEC son procesos graves con mortalidad muy elevada. La supervivencia a los 3 años de la enfermedad más característica de este grupo, la HAP idiopática, es del 47% sin tratamiento y de cerca del 70% si se trata con fármacos de última generación.
- 3.
La atención adecuada de los pacientes requiere de técnicas y procedimientos diagnósticos y terapéuticos complejos, disponibles sólo en centros específicos, que precisan para su realización de un grado elevado de especialización. Para poseer esta especialización es esencial disponer de experiencia, que sólo es posible alcanzar y mantener a través de un cierto volumen de actividad.
- 4.
Los fármacos específicos disponibles en la actualidad tienen un coste muy elevado, que, en régimen de monoterapia, oscila entre 460 y 23.000 € mensuales (tabla IV), y se administran crónicamente. Por ello, a fin de optimizar la relación coste-eficacia es preciso que la decisión terapéutica esté bien fundamentada y el tratamiento, estrechamente controlado.
- 5.
La concentración de casos en unidades de referencia debe hacer posible que éstas tengan acceso a un número crítico de pacientes que les permita participar en ensayos clínicos terapéuticos multicéntricos. Con ello, los pacientes pueden tener acceso a fármacos más eficaces y/o seguros antes de su comercialización. Asimismo, el sistema sanitario dispone de financiación externa de tratamientos cuyo coste es muy elevado.
- 1.
Personal:
- –
Mínimo de 2 médicos especialistas (neumólogos o cardiólogos) con interés en la enfermedad y experiencia profesional demostrable, que puedan cubrir las necesidades asistenciales de la unidad. Uno de ellos actuará como director o coordinador de la unidad y será su responsable.
- –
Mínimo de un diplomado/a en enfermería con especial dedicación a la unidad, que posea experiencia en la enfermedad y los distintos tratamientos empleados.
- –
Personal de apoyo administrativo para la coordinación asistencial de los pacientes.
La atención de los pacientes con HP tiene un marcado carácter multidisciplinario, por lo que la unidad debe actuar coordinadamente con los especialistas y unidades del centro que intervienen en los procesos diagnóstico y terapéutico.
- –
- 2.
Volumen de actividad. Atendiendo a las características de la organización sanitaria en España, el volumen de actividad mínimo exigible a una unidad de referencia en HP es de 5 casos nuevos de HAP al año, mantenido durante un mínimo de 3 años consecutivos, y al menos 30 pacientes en seguimiento clínico activo.
- 3.
Experiencia y calidad asistencial. Las unidades de referencia deben acreditar experiencia asistencial en los tipos más significativos de HP —HAP idiopática, HAP asociada a enfermedades del tejido conectivo, HAP asociada a infección por el VIH e HPTEC—, así como en las distintas formas de administración de fármacos, específicamente en la infusión continua por vía intravenosa. El personal de la unidad debe realizar sesiones clínicas regulares en las que se analice la evolución de los pacientes. Las unidades deben acreditar unos indicadores de resultados, específicamente de supervivencia, adecuados a los estándares actuales.
- 4.
Equipamiento y recursos disponibles. Las unidades de referencia en HP pertenecerán a centros terciaros que cuenten con el equipamiento y los recursos adecuados para atender a los pacientes. Al menos deben disponer de las siguientes unidades especializadas: ecocardiografía, hemodinámica cardíaca, laboratorio de función pulmonar, prueba de esfuerzo cardiopulmonar, unidad del sueño, servicio de radiodiagnóstico con capacidad para realizar angiografía por tomografía computarizada helicoidal y tomografía computarizada de alta resolución, medicina nuclear, unidad de cuidados intensivos y unidades de hospitalización de neumología y cardiología.
Además de las señaladas, también tiene especial interés que existan las siguientes unidades o equipos: trasplante pulmonar y/o cardiopulmonar, angiorradiología, unidad especializada en enfermedades autoinmunitarias (medicina interna, reumatología), unidad especializada en el VIH, servicios de cirugía cardíaca y de cirugía torácica, equipo quirúrgico experto en tromboendarterectomía pulmonar, trasplante hepático y/o unidad de hemodinámica hepática.
- 5.
Actuar de acuerdo con procedimientos normalizados de trabajo (PNT). La unidad debe disponer de PNT convenientemente actualizados de diagnóstico y tratamiento. En concreto, se requieren los siguientes: estudio hemodinámico y prueba vasodilatadora, ETT y ecocardiograma transesofágico, prueba de la marcha de 6 min, prueba de esfuerzo incremental, estudio radiológico de los pacientes con HPTEC, implantación de catéter intravenoso permanente, administración de fármacos por vía intravenosa, subcutánea e inhalada, manejo de la insuficiencia cardíaca derecha, admisión en unidades de cuidados intensivos, indicación de trasplante, actuación en estadio terminal.
Los PNT deben revisarse anualmente y analizar la validez diagnóstica de las pruebas realizadas, el grado de adecuación a las indicaciones previamente establecidas y la tasa de complicaciones en los procedimientos. Debería establecerse un calendario de auditoría externa de su cumplimento.
- 6.
Disponer de un sistema de información. Dicho sistema, preferentemente informatizado, debe permitir el conocimiento de la actividad y la evaluación de los resultados. Los pacientes deberían recogerse en una base de datos en la que consten los datos fundamentales de su diagnóstico y seguimiento terapéutico.
- 7.
Actividad investigadora. La unidad debe desarrollar actividad investigadora que permita profundizar en el conocimiento de la HP. Dicha actividad se concreta en el diseño y realización de proyectos de investigación propios, la participación en estudios clínicos nacionales e internacionales, la participación en registros y las publicaciones científicas.
- 1.
Consulta monográfica.
Concebida para el estudio inicial del paciente y el seguimiento evolutivo tras iniciar tratamiento. Para el correcto funcionamiento de la consulta se requiere: a) enfermería especializada en la enfermedad y entrenada en el funcionamiento de los dispositivos utilizados para la administración de fármacos (bombas de infusión, sistemas de inhalación, cuidados de la vía central permanente); b) red informática con acceso a la historia clínica, datos de laboratorio y exámenes realizados, y c) instrumental apropiado (negatoscopio, esfigmomanómetro, electrocardiógrafo, pulsioxímetro).
- 2.
Procedimientos diagnósticos.
La unidad debe tener a su alcance los recursos necesarios para establecer un diagnóstico específico de la clase y tipo de HP, de la situación hemodinámica, de la respuesta vasodilatadora y de la tolerancia al esfuerzo. Las unidades subespecializadas en HPTEC-tromboendarterectomía pulmonar deben disponer de medios para la localización de las lesiones trombóticas (angiografía pulmonar y angiografía por tomografía computarizada helicoidal).
- 3.
Procedimientos terapéuticos.
La unidad debe contar con los recursos adecuados para proporcionar los distintos tratamientos farmacológicos disponibles, en sus diferentes modalidades de administración. En concreto, debe disponer de medios para la implantación de acceso venoso central permanente para la administración de epoprostenol intravenoso, en caso de ser necesario, y de la organización necesaria para administrar este tratamiento: enfermería especializada, plan de formación del paciente y familiares, y plan de actuación ante complicaciones o efectos adversos.
En cuanto a los tratamientos no farmacológicos (septostomía interauricular, trasplante pulmonar o cardiopulmonar, tromboendarterectomía pulmonar), es de interés que en el centro haya unidades o equipos especializados en estos procedimientos, coordinados con la unidad de HP. En caso de no ser así, la unidad de HP debe tener protocolizados los criterios de derivación a los centros que cuenten con estas unidades especializadas y actuar de forma coordinada con ellas.
- 4.
Área de hospitalización.
Se requiere un área específica para el ingreso de los pacientes que precisen cuidados que no puedan prestarse de forma ambulatoria (necesidad de cuidados integrales, deterioro grave de la situación clínica) o para la realización de actuaciones diagnósticas o terapéuticas específicas. El ingreso puede sustituirse por una atención en el hospital de día en los pacientes en situación clínica estable.
- 5.
Atención permanente. Las unidades de referencia deben tener organizado un sistema de cobertura de 24 h, que garantice el acceso de los pacientes al cuidado especializado y que sea particularmente solvente en los problemas derivados de la utilización de los distintos dispositivos de administración de fármacos.
Habitualmente el diagnóstico inicial19 de sospecha de HP lo establecen médicos especialistas (cardiólogos, neumólogos, internistas o reumatólogos) del centro más próximo al paciente. Una vez establecida la sospecha diagnóstica, en determinados casos es preciso derivar al paciente a una unidad de referencia en hp. Las indicaciones para dicha derivación se especifican en la tabla IX.
Indicaciones para la derivación a una unidad de referencia en hipertensión pulmonar (HP)
Una vez excluida la cardiopatía izquierda como causa probable del aumento de la PSAP:
|
HPTEC: hipertensión pulmonar tromboembólica crónica; PSAP: presión sistólica arterial pulmonar.
El control y seguimiento del paciente ya diagnosticado debe efectuarse de forma coordinada entre la unidad clínica local y la de referencia. En la tabla X se resume brevemente la distribución de funciones entre ambas unidades.
Distribución de funciones entre la unidad clínica local y la unidad de referencia en hipertensión pulmonar
|
Es preciso que haya una comunicación fluida entre la unidad clínica local y la de referencia. Para ello se requiere:
- 1.
Informe completo de derivación del paciente de la unidad clínica local a la unidad de referencia.
- 2.
Informe de la unidad de referencia a la unidad clínica local en cada visita del paciente, que recoja el tratamiento actual y los problemas especiales que requieran una pauta específica de vigilancia.
- 3.
Facilitar a la unidad clínica local vías rápidas de comunicación con el personal de la unidad de referencia (teléfono, mensajería electrónica, fax).
- 4.
Garantizar la máxima celeridad en la evaluación diagnóstica y la instauración de tratamiento, especialmente en las situaciones de mayor gravedad.
- 5.
Dar a conocer a la unidad clínica local los protocolos básicos de diagnóstico y tratamiento de los pacientes con HP y acordar conjuntamente la forma óptima de cumplimiento.
- –
Dada la gravedad que conlleva el diagnóstico de HP, siempre que se sospeche ésta no debería demorarse la realización de la ETT, especialmente ante la sospecha de HAP o HPTEC, o cuando los síntomas sean acentuados (clases funcionales III o IV). En estas circunstancias la ETT debería considerarse preferente y no posponerse más de 4 semanas.
- –
Cuando se solicite la derivación de un paciente a una unidad de referencia en HP, no deberían transcurrir más de 3–4 semanas hasta la primera visita en dicha unidad, y otras 3–4 semanas hasta la realización del estudio hemodinámico diagnóstico.
- –
Una vez diagnosticado el tipo de HP y efectuada la evaluación hemodinámica, el tratamiento debería instaurarse en menos de 15 días.
- –
En pacientes estables y con buena evolución (clases funcionales I-II) las visitas clínicas de seguimiento pueden espaciarse a cada 3 meses en la unidad clínica local y cada 6 meses en la de referencia.
- –
En caso de deterioro clínico significativo o complicaciones graves del tratamiento, el paciente debe ser atendido en el centro de referencia en menos de 24 h.
En general, el paciente requiere seguimiento en una unidad de referencia hasta que se produce el fallecimiento o se realiza el trasplante. Los cuidados paliativos de los pacientes con HP en fases avanzadas e irreversibles de la enfermedad deben acordarse y realizarse de forma coordinada entre la unidad de referencia y la unidad clínica local.
7.2Situaciones especialesEn determinadas situaciones algunos pacientes con HP pueden requerir procedimientos para los que es preciso un alto grado de especialización. Dadas la particularidad de estas situaciones y la alta especialización requerida, no es necesario que estos procedimientos formen parte de las atribuciones propias de las unidades de referencia. Por este motivo, dichas unidades deben actuar de forma coordinada y tener protocolos de derivación preestablecidos con las unidades subespecializadas. Estas situaciones se plantean específicamente en las cardiopatías congénitas, el trasplante pulmonar o cardiopulmonar y la tromboendarterectomía pulmonar.
AgradecimientosLos miembros del grupo de trabajo agradecen la revisión del manuscrito y los comentarios efectuados por N. Galie y D. Jiménez.
Conflictos de interésA continuación se dan a conocer las relaciones de carácter financiero que los miembros del grupo de trabajo han mantenido durante los últimos 3 años con empresas farmacéuticas cuyos productos guardan relación directa o indirecta con el presente documento.
J.A. Barbera. Consultoría: Actelion, Encysive, Pfizer, Praxis. Conferencias y seminarios: Actelion, Ferrer, GSK, Pfizer, Schering. Ayudas a la investigación: Actelion, Schering. Ensayos clínicos: Actelion, Encysive, Lilly Icos, Lung RX, Myogen, Pfizer.
P. Escribano. Consultoría: GSK, Pfizer, Praxis. Conferencias y seminarios: Actelion, Ferrer, GSK, Pfizer, Schering. Ayudas a la investigación: Actelion, GSK. Ensayos clínicos: Actelion, Ferrer, Myogen, Pfizer.
P. Morales. Conferencias y seminarios: Pfizer, Schering.
M.A. Gómez. Consultoría: Ferrer, GSK, Pfizer, Praxis, Schering. Conferencias y seminarios: Actelion, Ferrer, GSK, Pfizer, Schering. Ayudas a la investigación: Actelion, GSK, Pfizer. Ensayos clínicos: Actelion, Gilead, Lilly Icos, Lung Rx, Pfizer, Schering, United Therapeutics.
M. Oribe. Conferencias y seminarios: Actelion, Ferrer, GSK, Pfizer, Schering.
A. Martínez. Consultoría: Pfizer, MSD, Schering. Ensayos: Astra, Ferrer, MSD, Schering, Sanofi-Aventis. Conferencias y seminarios: Almirall, BMS, Esteve, MSD, Pfizer, Sanofi-Aventis, Schering, Servier, Uriach. Ayudas a la investigación: Actelion, Pfizer.
A. Román. Consultoría: Actelion, Pfizer, Praxis. Conferencias y seminarios: Actelion, GSK, Pfizer, Schering. Ayudas a la investigación: Actelion. Ensayos clínicos: Actelion, Ferrer, Pfizer.
J. Segovia. Conferencias y seminarios: Actelion. F. Santos. Conferencias y seminarios: Actelion, Ferrer, GSK, Pfizer, Schering. Ayudas a la investigación: Ferrer, Pfizer. Ensayos clínicos: Pfizer.
M.T. Subirana. Consultoría: Actelion. Conferencias: Actelion, Pfizer. Ayudas a la investigación: Actelion.
Documento publicado simultáneamente en Rev Esp Cardiol. 2008;61(2):170-84.
La financiación necesaria para la elaboración de este documento se ha obtenido a través de ayudas no condicionadas concedidas a la SEPAR y la SEC por Actelion Pharmaceuticals España, Ferrer Grupo, GlaxoSmithKline, Pfizer y Schering España.
Coordinadores




www.publicationethics.org.




Archivos de Bronconeumología follows the Recommendations for the Conduct, Reporting, Editing and Publication of Scholarly Work in Medical Journals