


La combinación de fibrosis pulmonar y enfisema (CFPE), es un síndrome definido recientemente, en el cual coexisten en un mismo individuo enfisema en lóbulos superiores y fibrosis en lóbulos inferiores. Estos pacientes presentan un perfil funcional respiratorio característico, con volúmenes pulmonares dinámicos y estáticos aparentemente normales o mínimamente alterados que contrastan con una grave alteración de la difusión del monóxido de carbono (DLCO) e hipoxemia arterial, la cual empeora durante el esfuerzo. La prevalencia de hipertensión pulmonar es elevada y representa la principal condición que determina el pronóstico. La Tomografía axial computarizada de alta resolución (TCAR) constituye la herramienta primordial para confirmar su diagnóstico. Se ha postulado al humo del tabaco como el principal agente etiológico, sin embargo, ni los mecanismos fisiopatológicos ni la secuencia de eventos involucrados en este síndrome ha sido aún dilucidados. Estudios experimentales en modelos animales, están proporcionando información sobre la participación de algunos mediadores inflamatorios en su patogenia. Actualmente, no existe un consenso sobre la actitud terapéutica a seguir en estos pacientes, puesto que lo publicado hasta la fecha sobre esta entidad se limita a series de casos bien caracterizadas. Es por tanto, una patología con múltiples incógnitas todavía por resolver y con alta probabilidad de ser infradiagnosticada si no se tienen en cuenta sus particularidades clínico-funcionales.
The combination of pulmonary fibrosis and emphysema (CPFE) is a recently defined syndrome, in which anupper lobe emphysema and lower lobe fibrosis coexist in a single patient. These patients have a characteristic lung function profile, with dynamic and static lung volumes apparently normal or minimally altered, contrasting with a significant reduction of carbon monoxide transfer (DLco) and exercise hypoxemia. Pulmonary hypertension is highly prevalent and is the principal negative prognostic factor for this condition. High resolution computed axial tomography (HRCT) is the main tool to confirm the diagnosis. Cigarette smoking has been proposed as the main factor in its etiology; however, neither pathogenic mechanisms nor the sequence of events involved in this syndrome has been clarified yet. Experimental studies in animal models are providing information on the involvement of some inflammatory mediators in the pathogenesis. There is currently no consensus on the therapeutic approach to be followed in these patients, since those published to date on this subject are limited to well-characterised series of cases. Therefore, it is a pathology with many unknowns yet to be resolved and highly likely to be underdiagnosed, unless its functional clinical characteristics are taken into acount.
El enfisema pulmonar y las neumopatías intersticiales idiopáticas incluyendo la fibrosis pulmonar idiopática (FPI), son entidades definidas por distintos criterios clínicos, funcionales, radiológicos y patológicos1. La FPI, es la más común de las neumonías intersticiales idiopáticas, con peor pronóstico y mayor mortalidad, con una mediana de supervivencia de 3 años desde el momento del diagnóstico2. Su sustrato anatomopatológico es la neumonía intersticial usual (NIU) y se caracteriza por una alteración ventilatoria restrictiva y disminución en la transferencia de monóxido de carbono (DLCO). Por su parte, el enfisema se define como un agrandamiento de los espacios aéreos distales al bronquiolo terminal, debido a la destrucción de los tejidos que constituyen sus paredes3. El enfisema, es un fenómeno presente en la enfermedad pulmonar obstructiva crónica (EPOC), y que puede condicionar un patrón obstructivo por la limitación al flujo aéreo dada las diversas alteraciones estructurales que produce en el pulmón4–6.
La combinación de ambos procesos, aparentemente tan disímiles en la práctica clínica diaria, fue descrita por primera vez hace más de 30 años, por Auerbach7, en un estudio donde se analizaba anatomopatológicamente los pulmones de 1.824 autopsias. Ya en ese entonces, el referido autor sugirió el papel del tabaquismo como agente responsable de la coexistencia de estos hallazgos, basándose en estudios de modelos animales expuestos al humo del tabaco8,9. Con el advenimiento y desarrollo de la tomografia axial computarizada (TAC) en el arsenal diagnóstico de las enfermedades respiratorias, Wiggins et al10 correlacionaron a principio de los noventa, los hallazgos funcionales y radiológicos de 8 individuos con antecedentes de tabaquismo, quienes manifestaban disnea importante con espirometría sin evidencia de obstrucción, volúmenes estáticos conservados y DLCO intensamente alterada. Estos pacientes presentaban en la TAC áreas de enfisema de predominio en lóbulos superiores y lesiones compatibles con fibrosis pulmonar en ambas bases. Posteriormente, han sido publicadas varias series en la literatura1,10–22, la mayoría de ellas con metodología retrospectiva en el que hay que destacar la realizada por Cottin et al1 quien la caracteriza como una entidad clínica bien definida denominada «combinación de fibrosis pulmonar y enfisema» (CFPE).
Epidemiología y características clínicasLa prevalencia de la CFPE es desconocida, aunque se estima que podría representar entre un 5 y 10% de los casos de enfermedad pulmonar intersticial difusa23. Las cohortes estudiadas son en su gran mayoría hombres, siendo la naturaleza de este hallazgo hasta ahora, incierta15,16. Se presenta generalmente en mayores de 65 años, fumadores activos o exfumadores con historia de consumo elevado de tabaco (dosis acumulada de más de 40 paquetes/año). La exposición a compuestos agrícolas es otro dato epidemiológico que recogen algunas series1,21–24.
Desde el punto de vista clínico, la disnea de esfuerzo intensa (clase funcional iii o iv de la New York Heart Association) es el síntoma más comúnmente manifestado por los pacientes. En el examen físico, es usual encontrar dedos en palillo de tambor y crepitantes en «velcro» a la auscultación respiratoria en las bases pulmonares. Otros signos y síntomas reportados aunque con menor frecuencia han sido: tos, que puede ser seca o productiva, sibilantes, cianosis peribucal y astenia1,14,22,23.
La hipertensión pulmonar es una complicación frecuente y relevante en la historia natural de este síndrome1,15,23,25, puesto que su presencia condiciona un curso clínico peor y una menor supervivencia, como detallaremos más adelante. Por otro lado, varios autores han comunicado una prevalencia no despreciable de enfermedad vascular arterioesclerótica y carcinoma broncogénico en estos pacientes12,21,23. A pesar de que la concurrencia de estas enfermedades en la CFPE puede estar directamente relacionada con el tabaquismo como factor etiopatogénico común, hay algunos estudios que sugieren que la prevalencia de cáncer de pulmón puede variar en base al grupo étnico analizado, siendo las series japonesas las que demuestran una mayor frecuencia21, por lo cual es necesario disponer de estudios con un número amplio de pacientes para corroborar esta última observación.
PatogeniaA pesar de que el sustrato fisiopatológico de la CPFE es poco conocido, es de suponer que el mismo entraña un complejo proceso en el que se imbrican diversos tipos celulares, vías patogénicas comunes y mediadores con capacidad inflamatoria y/o fibrogénica, que conllevan finalmente a la destrucción del parénquima pulmonar y al remodelado aberrante característico de la fibrosis. A su vez, dichos mediadores celulares interactúan con factores ambientales que podrían intervenir como modificadores de la enfermedad ante un genotipo permisivo. A continuación, se repasarán los principales mecanismos etiopatogénicos descritos en relación con este síndrome.
EtiologíaSe ha sugerido como principal factor etiológico al humo del cigarrillo, dado que el antecedente de tabaquismo es un hecho constante en todas las cohortes reportadas10–23. El humo del tabaco, una mezcla compleja de más de 4.000 sustancias químicas, ha sido asociado a un largo espectro de enfermedades, siendo el enfisema, la alteración pulmonar más comúnmente atribuida a su consumo26. La caolinita o silicato de aluminio por ejemplo, es una sustancia inorgánica de uso industrial presente en el humo del tabaco y que ha sido aislada en los macrófagos alveolares de pacientes fumadores con enfisema pulmonar y con bronquiolitis respiratoria asociada a enfermedad intersticial (BR/EPID)27. Se ha postulado la hipótesis de que la acumulación de estos macrófagos mediada por la inhalación crónica de este agente, desencadena la serie de fenómenos fisiopatológicos que conducen finalmente al desarrollo de bronquiolitis respiratoria y enfisema28.
Por su parte, aunque la etiología de la FPI es desconocida, se piensa que sea consecuencia de la interacción de factores ambientales en individuos genéticamente predispuestos, siendo cada vez más reconocida la relación de de esta enfermedad con el tabaquismo, el cual produce también un efecto deletéreo demostrado sobre su pronóstico29,30. En este contexto, Katzenstein et al31 documentaron una inesperada frecuencia de fibrosis en piezas de resección pulmonar de pacientes fumadores candidatos a cirugía por neoplasia, sin que existiera evidencia clínica de enfermedad intersticial pulmonar. Cabe destacar, que el hábito de fumar ha sido asociado también con un patrón espirométrico restrictivo en amplios estudios epidemiológicos32, lo cual sustenta la hipótesis de que el tabaquismo es capaz de producir diversos tipos de afectación parenquimatosa pulmonar que se manifiestan en diferentes expresiones fenotípicas31.
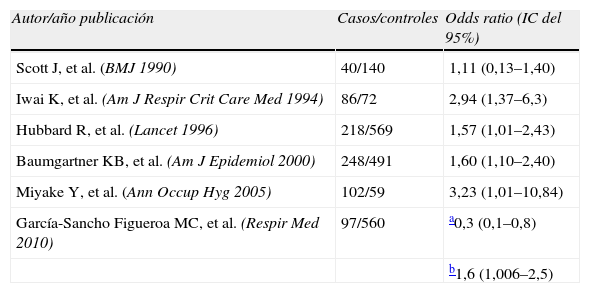
En la tabla 1 se enumeran los principales estudios que relacionan la exposición tabáquica y la FPI.
Estudios tipo caso-control realizados sobre exposición ambiental y fibrosis pulmonar idiopática. Odds ratio calculada para la exposición tabáquica
| Autor/año publicación | Casos/controles | Odds ratio (IC del 95%) |
| Scott J, et al. (BMJ 1990) | 40/140 | 1,11 (0,13–1,40) |
| Iwai K, et al. (Am J Respir Crit Care Med 1994) | 86/72 | 2,94 (1,37–6,3) |
| Hubbard R, et al. (Lancet 1996) | 218/569 | 1,57 (1,01–2,43) |
| Baumgartner KB, et al. (Am J Epidemiol 2000) | 248/491 | 1,60 (1,10–2,40) |
| Miyake Y, et al. (Ann Occup Hyg 2005) | 102/59 | 3,23 (1,01–10,84) |
| García-Sancho Figueroa MC, et al. (Respir Med 2010) | 97/560 | a0,3 (0,1–0,8) |
| b1,6 (1,006–2,5) |
IC: intervalo de confianza. Modificado de Taskar et al.30
La alta prevalencia de vasculopatía e hipertensión pulmonar descrita en los pacientes con CFPE, justifica también la imputación del cigarrillo en su génesis, puesto que el consumo del tabaco es un factor de riesgo bien conocido para el desarrollo de alteraciones vasculares. La exposición al humo del tabaco produce disfunción endotelial en las arterias coronarias y en las sistémicas. Se ha demostrado que la exposición de células endoteliales de arterias pulmonares al extracto de humo de tabaco, causa una inhibición irreversible de la actividad de la óxido nitrítico sintetasa endotelial (eNOS) debido a la reducción del contenido proteíco y del ARN mensajero. Esta inhibición de la actividad de la eNOS por el humo del tabaco puede explicar la disminución de su expresión en arterias pulmonares, observada en fumadores y puede predisponer a los pacientes a una mayor alteración de los vasos pulmonares33.
Stress oxidativoDentro de los mecanismos potencialmente implicados en el desarrollo de CFPE se ha postulado el aumento de estrés oxidativo y nitrosativo favorecido por el humo del tabaco, el cual aumenta la activación de células inflamatorias (leucocitos), que a su vez contribuirán al incremento, tanto local como sistémico, de los valores de oxidantes. El exceso de producción de oxidantes no neutralizado por los sistemas antioxidantes tisulares dan lugar a modificaciones estructurales a nivel de los tejidos epitelial, vascular y conectivo31.
MetaloproteinasasLas metaloproteinasas (MMP) constituyen una familia de enzimas producidas por células epiteliales alveolares, macrófagos y neutrófilos, las cuales actúan en el desarrollo del enfisema, gracias a su marcada actividad proteolítica y su capacidad para degradar el colágeno. La expression de la actividad de las MMP es modulada por diversas citoquinas, entre ellas la interleucina 13 (IL-13), la cual es capaz de producir en modelos experimentales, alargamiento de los espacios aéreos, fibrosis y remodelado inflamatorio en las vías aéreas34. En un estudio reciente en un subgrupo de pacientes con CFPE, se observó un aumento de expresión de algunas MMP en las áreas de afectación combinada de enfisema y NIU, lo cual sugiere que estas proteínas también contribuyen al depósito de matriz extracelular y al remodelado tisular anómalo, característico del proceso fibrótico26.
Caveolina-1Las caveolas son invaginaciones de la membrana celular presentes en varios tipos celulares. Estas estructuras son ricas en proteínas, así como en lípidos y colesterol y tanto su formación como su mantenimiento se deben principalmente a unas proteínas denominadas caveolinas, siendo la caveolina 1 la más estudiada. Esta proteína es un potente inmunomodulador, a la cual se le adscriben varias funciones, entre ellas, la transducción de señales, mediación de la apotopsis celular, regulación de la homeostasis del calcio intracelular, de la eNOs y supresión de tumores35,36. El tejido pulmonar expresa altos niveles de caveolinas, por lo que su disfunción se ha relacionado con la biopatología de la hipertensión pulmonar asociada a la EPOC, al enfisema, fibrosis intersticial y cáncer de pulmón. Por lo tanto, la caveolina-1 puede ser otro mediador importante en el origen de la CFPE, la cual sin duda, será merecedor de futuras investigaciones.
Modelos animalesSe han realizado tres estudios en modelos animales que han logrado reproducir los elementos histopatológicos de la CFPE. El primero, llevado a cabo por Hoyle et al37, demuestra que la sobreexpresión del factor de crecimiento derivado de las plaquetas (PDGF), produjo alargamiento de los espacio aéreos, inflamación y fibrosis en pulmones de ratones transgénicos. El PDGF es un factor de crecimiento con efecto pleiotrópico sobre varias estirpes celulares y que ha sido implicado tanto en la patogénesis de la FPI como en la del enfisema. Esta acción dicotómica se puede fundamentar en su actividad mitógena sobre los fibroblastos, su interacción con diversos marcadores inflamatorios y su probable capacidad de inducir un desequilibrio proteasa/antiproteasa en la matriz extracelular que propiciaría el desarrollo de enfisema.
La sobrexpresión tisular pulmonar de otros mediadores como el factor de necrosis tumoral alfa (TNF-α) y del factor transformador del crecimiento beta (TGF-β), han sido también capaces de generar este complejo fenotipo de fibrosis y enfisema en animales de experimentación38,39. El TNF-α es una citocina multifuncional que parece tener algunas actividades profibróticas40 y es considerado un mediador fundamental en distintas manifestaciones pulmonares y sistémicas que ocurren en diversas enfermedades respiratorias. El cigarrillo causa liberación de esta citocina en los pulmones, tanto en humanos como en modelos animales y sus niveles se han encontrado aumentados en el esputo y en sangre periférica de los pacientes con EPOC41.
Por su parte, el TGF-β es uno de los mediadores profibrogénicos más potentes con reconocida relevancia en la patogénesis de la FPI. La isoforma TGF-β1, induce la diferenciación de fibroblastos a miofibroblastos, la transición de células epiteliales a fibroblastos y la síntesis de moléculas de matriz extracelular, además de favorecer la apoptosis de células del epitelio alveolar40. Su papel en el desarrollo de enfisema es menos claro, sin embargo se ha observado un aumento de su expresión en el epitelio bronquiolar y en los macrófagos de las pequeñas vías aéreas de pacientes con EPOC42. En definitiva, el TGF-β es importante en la transición que hay desde la respuesta inflamatoria inmunitaria hasta el proceso de remodelación tisular43,44.
DiagnósticoPruebas de imagenEn la radiografía de tórax simple se puede apreciar un patrón intersticial o infiltrados reticulonodulares de distribución bibasal y periférica (subpleural) e hiperclaridad en los vértices con disminución de la trama vascular a ese nivel (fig. 1). Sin embargo, la radiografía de tórax puede inadvertir los hallazgos radiológicos de la CFPE, siendo la TCAR la exploración de referencia para confirmar el diagnóstico23,45. Las imágenes muestran la presencia de enfisema de predominio en lóbulos superiores, definido por una zona hipodensa bien delimitada sin pared, o con una pared muy fina y/o múltiples bullas, que coexiste con diversas manifestaciones radiológicas propias de las enfermedades difusas del pulmón, como lo son opacidades reticulares, bronquiectasias de tracción, áreas de engrosamiento septal, vidrio deslustrado y panal de abeja en lóbulos inferiores (fig. 2). Las lesiones enfisematosas corresponden a enfisema centrolobulillar y paraseptal (bullas subpleurales). El enfisema paraseptal se ha descrito hasta en una 90% de los casos, por lo que algunos autores sugieren que se trata de un rasgo característico de la CFPE1,23,46.

Radiografía de tórax de un paciente diagnosticado de combinación de fibrosis pulmonar y enfisema. Se observa afectación patrón intersticial bilateral, de predominio derecho con infiltrados reticulonodulares de distribución basal y subpleural y disminución de la densidad pulmonar en campos superiores, principalmente izquierdo.
 del mismo paciente. A) Presencia de enfisema paraseptal y bullas subpleurales (cabezas de flecha blancas) y enfisema centrolobulillar (flechas) en ambos lóbulos superiores. B) Afectación intersticial reticular con engrosamiento intralobulillar e imágenes de panalización subpleural y bronquiectasia de tracción (cabezas de flecha negras) C) Afectación intersticial reticular en lóbulo medio y lóbulo inferior derecho , con engrosamiento de septos interlobulillares, panalización subpleural y bronquiectasias por tracción. D) Reconstrucción coronal a nivel de regiones posteriores de ambos pulmones: Enfisema paraseptal bilateral (cabezas de flecha blancas) y afectación intersticial reticular y panalización en lóbulo inferior derecho.")
Tomografía computarizada de alta resolución (TCAR) del mismo paciente. A) Presencia de enfisema paraseptal y bullas subpleurales (cabezas de flecha blancas) y enfisema centrolobulillar (flechas) en ambos lóbulos superiores. B) Afectación intersticial reticular con engrosamiento intralobulillar e imágenes de panalización subpleural y bronquiectasia de tracción (cabezas de flecha negras) C) Afectación intersticial reticular en lóbulo medio y lóbulo inferior derecho , con engrosamiento de septos interlobulillares, panalización subpleural y bronquiectasias por tracción. D) Reconstrucción coronal a nivel de regiones posteriores de ambos pulmones: Enfisema paraseptal bilateral (cabezas de flecha blancas) y afectación intersticial reticular y panalización en lóbulo inferior derecho.
En algunos casos, la delimitación entre las imágenes de enfisema y de fibrosis es compleja, observándose un área de transición entre ambas zonas que dificulta su correcta interpretación46. La riqueza de la semiología radiológica en la TCAR de estos pacientes, se correlaciona estrechamente con los datos histopatológicos, siendo la NIU el patrón descrito con mayor frecuencia, sin embargo también se ha comunicado lesiones compatibles con neumonía intersticial no usual, y neumonía intersticial descamativa16, siendo esta última una patología ligada al consumo de tabaco.
Función pulmonarLa coexistencia de enfisema y fibrosis determina un perfil funcional característico que contrasta con el grado de disnea que manifiestan estos pacientes. La capacidad vital forzada (FVC), el volumen espiratorio forzado en el primer segundo (FEV1) y la capacidad pulmonar total (TLC), suelen encontrarse en el rango de la normalidad o poco alterados, a diferencia de la DLCO que se encuentra gravemente disminuida. La hipoxemia es un hallazgo frecuente, generalmente de grado moderado durante el reposo y que empeora durante el ejercicio1,18,23. La presencia de hiperinsuflación con aumento de la compliance pulmonar por pérdida de elasticidad en las áreas enfisematosas probablemente compense la pérdida de volumen debida a la fibrosis17–19. Por el contrario, la superposición de ambas patologías podría tener efectos sinérgicos deletéreos sobre el intercambio gaseoso que resultan en un descenso intenso de la DLCO. Esta particular alteración funcional lleva al menos a dos importantes repercusiones clínicas: la primera es que la presencia de volúmenes normales no excluye el diagnóstico de fibrosis pulmonar en este tipo de pacientes y la segunda es que ni la FVC ni la TLC pueden ser utilizadas como parámetros de seguimiento de la enfermedad debido a que no reflejan el grado de compromiso funcional. En este caso, la DLCO sería la variable que mejor se correlaciona con el grado de destrucción del parénquima. No obstante, una baja DLCO también puede ser indicativa de alteraciones en el lecho vascular pulmonar, en concreto, de hipertensión pulmonar (HP), puesto que se trata de una condición altamente prevalente en esta entidad.
Hipertensión pulmonarLa HP es una complicación frecuente en el curso clínico de la CFPE y es la principal condición que influye en su evolución y pronóstico23. Se ha comunicado una prevalencia en estos pacientes que oscila entre el 47% y el 90% mucho más elevada que en la EPOC o en la FPI aislada47. En la mayoría de las series publicadas, el diagnóstico de HP ha sido establecido mediante ecocardiografía transtorácica, siendo el criterio para definirla una presión sistólica en la arteria pulmonar (PSAP) estimada en ≥45mmHg. En el estudio de Cottin et al1, la presencia de HP fue predictor independiente de mortalidad, con un hazard ratio de 4,03; (p=0,03). La probabilidad de supervivencia a los 5 años era de 25% en los pacientes con HP demostrada por ecocardiograma frente a un 75% en aquellos sin evidencia de HP en el momento del diagnóstico. La mediana de supervivencia en esta serie fue de 6.1 años y se reducía a 3.9 años en aquellos con HP asociada.
En este sentido, Mejía et al15 publicó recientemente un estudio en el cual se comparaba diferentes variables clínicas, funcionales y pronosticas sobre un grupo pacientes con CFPE y pacientes con FPI sin evidencia de enfisema. Mediante un modelo de regresión logística se puso de manifiesto que una PSAP ≥ a 75mmHg era junto con la FVC una de las principales variables que determinaba la supervivencia, la cual fue menor en el grupo con CFPE. En estos pacientes, la extensión del enfisema establecida por TCAR se correlacionó positivamente con la PSAP.
La importancia de la hemodinámica pulmonar sobre la mortalidad en esta entidad, ha sido evidenciada en otro trabajo realizado también por el grupo de Cottin25 sobre 40 pacientes con CFPE, quienes tenían HP confirmada mediante cateterismo cardíaco derecho. Los factores que se asociaron con un peor pronóstico fueron: resistencia vascular pulmonar aumentada, índice cardíaco disminuido, frecuencia cardiaca elevada y DLCO baja. En este estudio, el diagnóstico de HP se estableció con una media de 16 meses luego del diagnóstico inicial de CFPE, y la tasa estimada de supervivencia al año de esta cohorte fue de 60%.
Por tanto, el cribado sistemático de HP en estos pacientes mediante ecocardiografía doppler se justificaría dada su alta prevalencia y su importante papel en el pronóstico. La presencia de enfisema puede dificultar su interpretación por lo cual algunos autores proponen valorar métodos diagnósticos alternativos como la resonancia magnética (RM). La técnica de flujo por RM ofrece una buena correlación con los parámetros hemodinámicos48,49, sin embargo, el acceso y el uso generalizado de esta exploración en este tipo de pacientes se encuentra aún muy restringido.
TratamientoLas opciones terapéuticas en la CFPE son limitadas. El tratamiento con corticoides sistémicos e inmunosupresores se ha utilizado a modo de analogía con el tratamiento de la FPI, sin haber demostrado claros beneficios en la series publicadas. La supresión completa del tabaquismo es una medida razonable que probablemente, evite la progresión de las lesiones enfisematosas. La posibilidad de utilizar la terapia específica aprobada para el manejo de la hipertensión arterial pulmonar (antagonistas de los receptores de endotelina 1, prostanoides o inhibidores de la fosfodiesterasa de tipo 5), tal y como se ha ensayado en la EPOC o en la FPI ha sido considerada por algunos autores, sin embargo, hasta la fecha no se dispone de ningún estudio publicado al respecto. Es importante señalar, que en estos pacientes la presencia de enfisema y de alteraciones en el lecho vascular pulmonar puede asociarse a un desequilibrio en la relaciones ventilación-perfusión (V/Q), siendo la vasoconstricción hipóxica uno de los principales mecanismos para evitar el agravamiento de la hipoxemia. Estos fármacos vasodilatadores pueden empeorar la oxigenación arterial al inhibir este mecanismo, por lo cual es necesario realizar estudios adecuadamente diseñados que permitan evaluar de forma detallada, el efecto de los mismos sobre el intercambio gaseoso.
ConclusionesLa coexistencia de fibrosis pulmonar y enfisema, comporta unos hallazgos clínicos, funcionales y radiológicos a primera vista contradictorios, que pueden confundir al clínico y conllevar a su infradiagnóstico. La presencia de HP asociada es una complicación frecuente, importante de identificar, ya que determina su pronóstico. El solapamiento de la amplia variedad de lesiones radiológicas e histopatológicas descritas en esta entidad, pone en evidencia la capacidad del tabaco como principal agente implicado, de generar diversas alteraciones parenquimatosas con diferentes expresiones fenotípicas. Una mejor comprensión de su fisiopatología y de los mecanismos moleculares involucrados, permitirá el desarrollo de futuras estrategias terapéuticas, sin olvidar que la prevención y el tratamiento del tabaquismo, sean posiblemente, las intervenciones que mayor impacto tendrán sobre su historia natural.
Al Dr. Ignasi Guasch por su continuo soporte y la cesión de las imágenes radiológicas.







