


El pseudotumor inflamatorio es una entidad rara que supone menos del 1% de todos los tumores localizados en el pulmón1. Refleja características radiológicas superponibles en las técnicas tomográficas convencionales respecto a otras entidades2, lo cual dificulta el diagnóstico diferencial, especialmente con el cáncer de pulmón.
Se presenta el caso de una mujer de 49 años, exfumadora, con un índice de consumo acumulado de 2 paquetes/año, con antecedentes personales de hipotiroidismo. Como tratamiento habitual tenía levotiroxina de 75mcg, un comprimido al día. Acudió a urgencias hospitalarias por un cuadro de cefalea tras un episodio de sinusitis, sin clínica respiratoria asociada, donde en el estudio complementario básico se incluyó una radiografía simple de tórax, en la que se visualizó una lesión apical derecha de contornos irregulares, como hallazgo incidental. Se solicitó una primera tomografía axial computarizada que fue informada como «masa tumoral redondeada en el vértice pulmonar derecho, de contornos irregulares, lobulados y con espiculaciones hacia el parénquima circundante. La lesión presenta un realce heterogéneo con el contraste, contactando ampliamente con la pleura apical posterior y superior, con un tamaño aproximado de unos 45×42mm, sin evidencia de extensión o infiltración pleural o extrapleural; en el vértice pulmonar izquierdo se visualiza una pequeña imagen nodular subcentimétrica, subpleural, de dudosa significación». Ante este hallazgo se realizó una tomografía por emisión de positrones donde se destacó la presencia de dicha masa, metabólicamente activa, con un valor de consumo entandarizado máximo (SUVmáx) de 27,5, y la observada en el lóbulo superior izquierdo con un SUVmáx de 7,5. A nivel mediastínico se informó de adenopatías hiliares bilaterales, destacando un conglomerado hiliar izquierdo de 3,2×2,1×4,1cm con un SUVmáx de 14,1 (fig. 1). Ante la alta sospecha de malignidad se decidió la realización de una biopsia con aguja gruesa de la lesión mayor, cuyo resultado mostró la presencia de fibrosis e infiltrado linfohistiocitario polimorfo sugestivo de proceso inflamatorio o pseudotumor inflamatorio con estudio molecular negativo para mutaciones del gen EGFR. No se realizó estudio para el gen ALK. Los marcadores tumorales (CEA, CA 15.3, CA 19.9, CYFRA-21, Ag de células escamosas, Pro-GAP y enolasa) fueron negativos. No presentó anemia ni trombocitosis. Tras este resultado se estimó necesaria la confirmación mediante la realización de una nueva biopsia con aguja gruesa, cuyo diagnóstico fue similar y la realización de una fibrobroncoscopia, en la que se visualizó una mucosa engrosada de aspecto inflamatorio en el lóbulo inferior izquierdo, de donde se realizó cepillado bronquial a ciegas sin obtención de células neoplásicas. Ante la ausencia de datos de malignidad y la existencia de varias lesiones que dificultaban el abordaje quirúrgico, se acordó el inicio de tratamiento con deflazacor a dosis de 1mg/kg de peso al día durante 2 meses, reduciéndose a la mitad los siguientes 2 meses y suspendiéndose al mes siguiente. Se realizaron 3 estudios de tomografía axial computarizada durante el tratamiento, observándose una disminución progresiva del tamaño tumoral, así como la presencia de tractos lineales fibrótico-cicatriciales y bronquiectasias por tracción en la zona, con desaparición del nódulo contralateral.
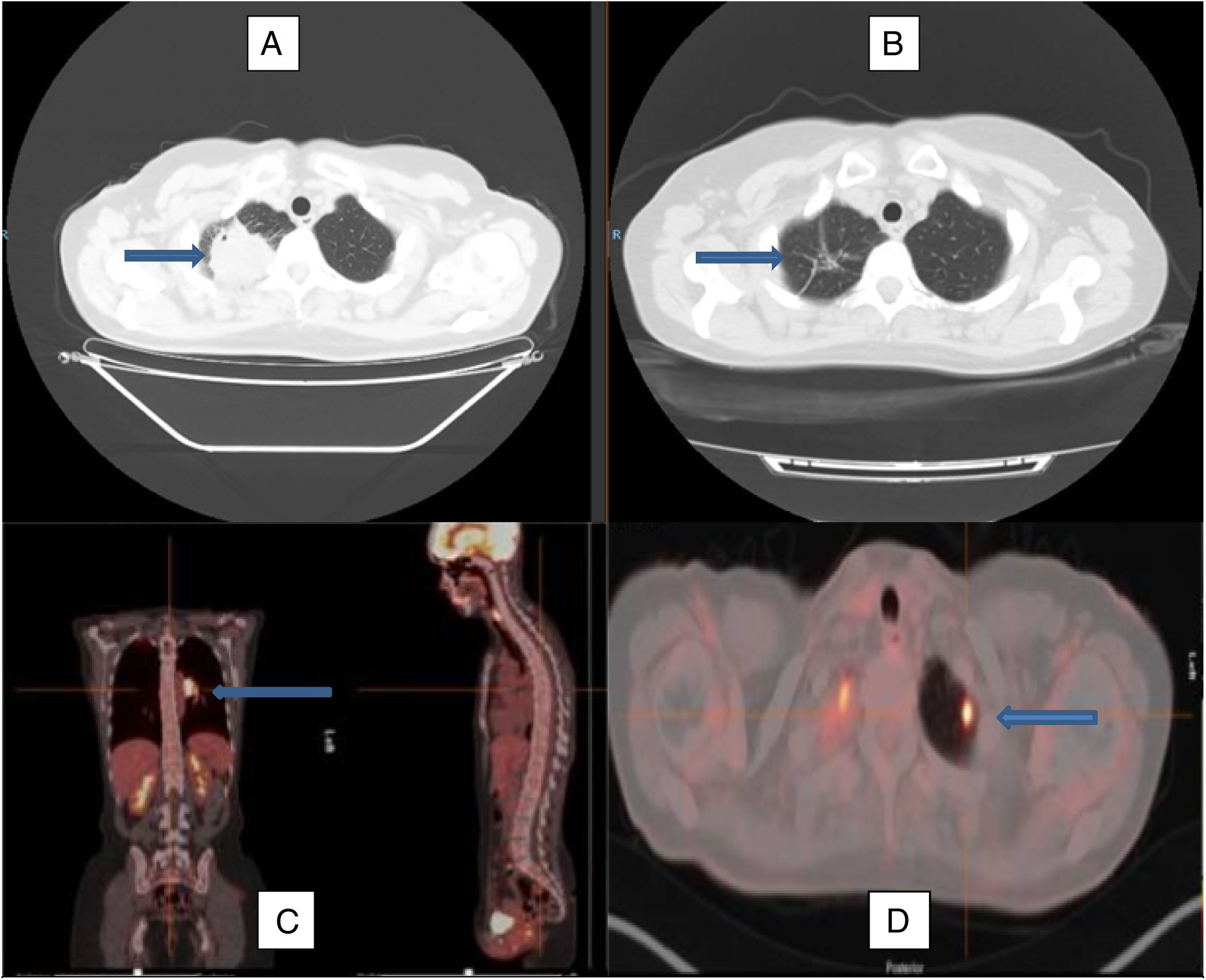
Imagen de tomografía axial computarizada al diagnóstico (A) y tras el tratamiento con corticoides (B), observándose la remisión de la lesión en el lóbulo superior derecho, permaneciendo en su localización algunos tractos fibrosos residuales (flechas). Imagen de PET-TAC con imagen hipercaptante en el hilio pulmonar izquierdo (C) y en el lóbulo superior izquierdo (D), marcadas por las flechas, que reflejan la afectación multilocular del caso.
El pseudotumor inflamatorio puede localizarse en cualquier órgano, pero afecta principalmente al pulmón1 y suele ser más frecuente en las 2 primeras décadas de la vida2. Normalmente se presenta como una masa única, circunscrita, mayor de 3cm y más frecuentemente en los lóbulos inferiores3. Puede aparecer en la bibliografía con distintas denominaciones (granuloma de células plasmáticas, proliferación o tumor miofibroblástico inflamatorio, histiocitoma, xantoma, fibroxantoma, xantogranuloma, xantoma fibroso, pseudotumor xantomatoso, plasmocitoma, granuloma solitario de mastocitos, fibrosarcoma inflamatorio)4, lo que da cuenta de la complejidad, variabilidad histológica y comportamiento de esta entidad, hasta el punto de que, en ocasiones, puede ser clasificado como una verdadera neoplasia5. Su etiología es desconocida, aunque se cree que puede haber varios factores predisponentes: cirugías, traumatismos, reacciones inmunológicas, esteroides, radioterapia o infecciones6. El papel de un agente infeccioso parece quedar restringido a los primeros estadios, iniciándose así una cascada de reacciones, a través de las cuales el tumor llega a ser autónomo7. Entre los microorganismos más frecuentemente asociados se incluyen las micobacterias, el virus de Epstein-Barr, Actinomycetos y Mycoplasma y se han descito algunos casos relacionados con Corynebacterium equi, Escherichia coli, Klebsiella, Bacillus sphaericus, Pseudomonas, Helicobacter pylori, Coxiellaburnetti4,8,9, virus del herpes simple e incluso con infección por el VIH10. La anatomía patológica típica muestra bandas fibrosas interrumpidas por láminas irregulares de linfocitos, histiocitos y células plasmáticas policlonales11. Aunque la resección quirúrgica es el tratamiento de elección12, en los últimos años, en varios estudios, se ha documentado que en aproximadamente la mitad de los casos se presentan reordenamientos del gen ALK en el cromosoma 2p23, lo que causa su expresión aberrante13. Esto sugiere que este subgrupo podría mostrar sensibilidad a los inhibidores de la tirosina quinasa como el crizotinib14. Solamente existen series de casos publicados que apoyan su eficacia6,13, y ningún ensayo que compare la administración de crizotinib frente a corticoides en este subgrupo.
La terapia corticoidea presenta una eficacia variable con resultados limitados y no concluyentes en la literatura1,12,14,15, siendo utilizada en primera línea en pacientes no candidados a cirugía1,14,15, como en la afectación bilateral. Lee et al. reportaron un caso similar en el que la respuesta a corticoides fue excelente, sin mostrar evidencia de recurrencia tras 20 meses de seguimiento tras la desaparición de las lesiones14. Díez et al. obtuvieron resultados similares tras tratamiento corticoideo a dosis bajas después de 30 meses15. Son pocos los casos descritos con lesiones bilaterales como el presentado y se desconocen la eficacia y el índice de recurrencias tras el tratamiento esteroideo, por ausencia de estudios al respecto.







