



El déficit hereditario de la alfa-1 antitripsina (AAT) se puede manifestar clínicamente como una enfermedad pulmonar obstructiva crónica (EPOC). Se define por una concentración sérica por debajo del 35% del valor medio esperado, o 50mg/dL (medida por nefelometría) y está relacionado en más del 95% de los casos, con genotipos Pi*ZZ, y muy infrecuentemente con otros genotipos resultantes de combinaciones entre alelos Z, S, raros y nulos. Se ha realizado una revisión sistemática cualitativa de 107 artículos, centrados principalmente en la búsqueda activa del déficit de AAT (DAAT) en pacientes con EPOC y en el tratamiento con AAT por vía intravenosa (iv). El comité asesor del Registro Español de pacientes con DAAT, sobre la base de esta revisión, considera que se debe descartar el DAAT, mediante la cuantificación de las concentraciones séricas de AAT, en todos los pacientes con EPOC y cuando sean bajas se debe completar el estudio mediante la determinación del fenotipo y, en ocasiones, del genotipo. El tratamiento de los individuos con EPOC asociado a DAAT grave debe incluir el tratamiento farmacológico y no farmacológico recomendado en las normativas de la EPOC. Existe suficiente evidencia, derivada de grandes estudios observacionales y de ensayos clínicos aleatorizados con placebo, que demuestran que el tratamiento con AAT iv disminuye la mortalidad y reduce la velocidad de progresión del enfisema, por lo que está indicado en casos seleccionados que cumplan los criterios de inclusión establecidos en las normativas internacionales.
La terapia con infusiones iv periódicas de AAT es el único tratamiento específico que existe para frenar la progresión del enfisema asociado al DAAT.
The effect of hereditary alpha-1 antitrypsin (AAT) deficiency can manifest clinically in the form of chronic obstructive pulmonary disease (COPD). AAT deficiency (AATD) is defined as a serum concentration lower than 35% of the expected mean value or 50mg/dl (determined by nephelometry). It is associated in over 95% of cases with Pi*ZZ genotypes, and much less frequently with other genotypes resulting from combinations of Z, S, rare and null alleles. A systematic qualitative review was made of 107 articles, focusing mainly on an active search for AATD in COPD patients and intravenous (iv) treatment with AAT. On the basis of this review, the consultant committee of the Spanish Registry of Patients with AATD recommends that all COPD patients be screened for AATD with the determination of AAT serum concentrations, and when these are low, the evaluation must be completed with phenotyping and, on occasions, genotyping. Patients with severe AATD COPD should receive the pharmacological and non-pharmacological treatment recommended in the COPD guidelines. There is enough evidence from large observational studies and randomized placebo-controlled clinical trials to show that the administration of iv AAT reduces mortality and slows the progression of emphysema, hence its indication in selected cases that meet the inclusion criteria stipulated in international guidelines.
The administration of periodic infusions of AAT is the only specific treatment for delaying the progression of emphysema associated with AATD.
El déficit hereditario de la alfa-1 antitripsina (AAT) se puede manifestar clínicamente como EPOC (típicamente enfisema pulmonar panacinar), cirrosis hepática en cualquier edad y, con menor frecuencia, como paniculitis, vasculitis sistémicas y otras enfermedades1. El déficit de AAT (DAAT) grave, definido por una concentración sérica inferior al 35% del valor medio esperado, o 50mg/dL (medida por nefelometría), está relacionado en más del 95% de los casos con genotipos Pi*ZZ, y muy infrecuentemente con otros genotipos resultantes de combinaciones entre alelos Z, S, raros y nulos2.
Dado que la detección de casos con DAAT grave conlleva la puesta en práctica de consejo genético, el estudio de familiares consanguíneos y, en casos seleccionados, la administración de infusiones iv periódicas de AAT, en el año 2006 la Sociedad Española de Neumología y Cirugía Torácica (SEPAR), en colaboración con el comité asesor del Registro Español de Pacientes con Déficit de AAT (REDAAT), editó una normativa sobre el diagnóstico y el tratamiento del DAAT, cuyos conceptos básicos siguen vigentes en la actualidad3. Sin embargo, diversos estudios posteriores4–7 han aportado nuevos datos que avalan la importancia de la detección del DAAT en los individuos con EPOC y la prescripción de tratamiento con AAT por vía iv en los pacientes con EPOC y DAAT grave8–13, todo lo cual justifica la presente actualización.
MetodologíaLos autores realizaron una búsqueda bibliográfica de artículos publicados entre 1985 y 2013 en las bases de datos Medline, Embase y la Cochrane Library, utilizando las palabras clave: «alpha-1 antitrypsin deficiency», «COPD», «asthma», «bronchiectasis», «augmentation therapy» y «replacement therapy». Los metaanálisis y las revisiones sistemáticas de otros autores, fundamentados en grados de evidencia científica, así como algunos artículos, citados en los seleccionados previamente y no detectados en las bases de datos, fueron asimismo incluidos para análisis.
Con los términos «alpha-1 antitrypsin deficiency» y «COPD» se obtuvieron 289 resúmenes; con «alpha-1 antitrypsin deficiency» y «asthma», 154; con «alpha-1 antitrypsin deficiency» y «bronchiectasis», 87; con «alpha-1 antitrypsin deficiency» y «augmentation therapy», 129; con «alpha-1 antitrypsin deficiency» y «replacement therapy», 71.
Después de 3 reuniones generales y una final monográfica, los autores de este artículo realizaron un análisis sistemático cualitativo de los artículos seleccionados para la elaboración de este documento. Tras eliminar los repetidos en las diferentes búsquedas, y a partir de la información proporcionada en el resumen de los seleccionados (o cuando este no era lo bastante explícito, del texto completo), se escogieron, por consenso, 107 artículos1–107, la mayoría centrados en la búsqueda activa del DAAT en pacientes EPOC y en el tratamiento con AAT por vía iv en pacientes con EPOC asociada a DAAT grave. Los autores valoraron individualmente los manuscritos considerados potencialmente útiles y los calificaron siguiendo los criterios de la Clasificación de las recomendaciones y calidad de evidencia según el Sistema Grade y del Reglamento para la Redacción de Normativas SEPAR108,109 y los criterios de la American College of Chest Physicians Task Force110, modificados por el Canadian Thoracic Society COPD Clinical Assembly Alpha-1 Antitrypsin Deficiency Expert Working Group13. Tras la revisión de los resultados, se consensuaron las conclusiones descritas a continuación, entre los miembros del comité asesor.
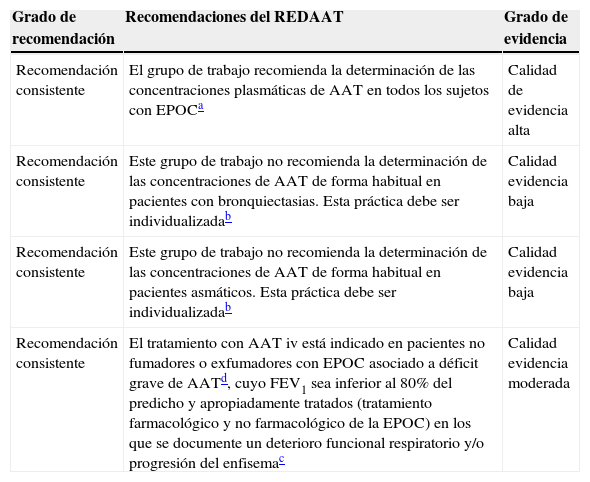
ResultadosLos resultados del análisis sistemático cualitativo aparecen resumidos en las tablas 1 y 2. El grupo de trabajo del REDAAT debe dejar constancia de la detección de importantes carencias en la bibliografía, que ponen de manifiesto la necesidad de llevar a cabo en el futuro estudios de alta calidad para responder apropiadamente a varias de las cuestiones planteadas. Aún así, el análisis de 4 trabajos seleccionados49,54,71,93 y un metaanálisis reciente de alta calidad13, centrados en la investigación del DAAT en la EPOC, permite afirmar que se debe descartar el DAAT, mediante la determinación de las concentraciones séricas de AAT, en todos los pacientes con EPOC y, en los que estas sean bajas, se debe completar el estudio mediante la obtención del fenotipo y, en ocasiones, del genotipo (recomendación consistente con calidad de evidencia alta, y que confirma lo propuesto en la normativa del 2006)3.
Resumen de las recomendaciones del REDAAT sobre el cribado del DAAT en EPOC, bronquiectasias y asma bronquial, y sobre aplicación de tratamiento con AAT iv
| Grado de recomendación | Recomendaciones del REDAAT | Grado de evidencia |
|---|---|---|
| Recomendación consistente | El grupo de trabajo recomienda la determinación de las concentraciones plasmáticas de AAT en todos los sujetos con EPOCa | Calidad de evidencia alta |
| Recomendación consistente | Este grupo de trabajo no recomienda la determinación de las concentraciones de AAT de forma habitual en pacientes con bronquiectasias. Esta práctica debe ser individualizadab | Calidad evidencia baja |
| Recomendación consistente | Este grupo de trabajo no recomienda la determinación de las concentraciones de AAT de forma habitual en pacientes asmáticos. Esta práctica debe ser individualizadab | Calidad evidencia baja |
| Recomendación consistente | El tratamiento con AAT iv está indicado en pacientes no fumadores o exfumadores con EPOC asociado a déficit grave de AATd, cuyo FEV1 sea inferior al 80% del predicho y apropiadamente tratados (tratamiento farmacológico y no farmacológico de la EPOC) en los que se documente un deterioro funcional respiratorio y/o progresión del enfisemac | Calidad evidencia moderada |
AAT: alfa-1 antitripsina; iv: intravenosa; DAAT: déficit de alfa-1 antitripsina; EPOC: enfermedad pulmonar obstructiva crónica; FEV1: volumen forzado espirado en el primer segundo; REDAAT: Registro Español de Pacientes con Déficit de Alfa-1 Antitripsina.
Recomendación consistente, calidad de evidencia alta. Puede aplicarse a la mayoría de pacientes en la mayoría de circunstancias.
Recomendación consistente, calidad evidencia baja. Puede cambiar cuando se disponga de otras pruebas.
Recomendación consistente, calidad evidencia moderada. Puede cambiar cuando se disponga de otras pruebas.
El déficit grave se define por concentraciones séricas de AAT≤50mg/dL, medidos por nefelometría. Generalmente, se asocia a fenotipos PIZZ y combinaciones de alelos «raros» y «nulos» entre sí o con Z y S. No se considera déficit grave al asociado a los fenotipos MZ, ni a la mayoría de los SZ, con excepción de aquellos que presenten concentraciones de AAT≤50mg/dL.
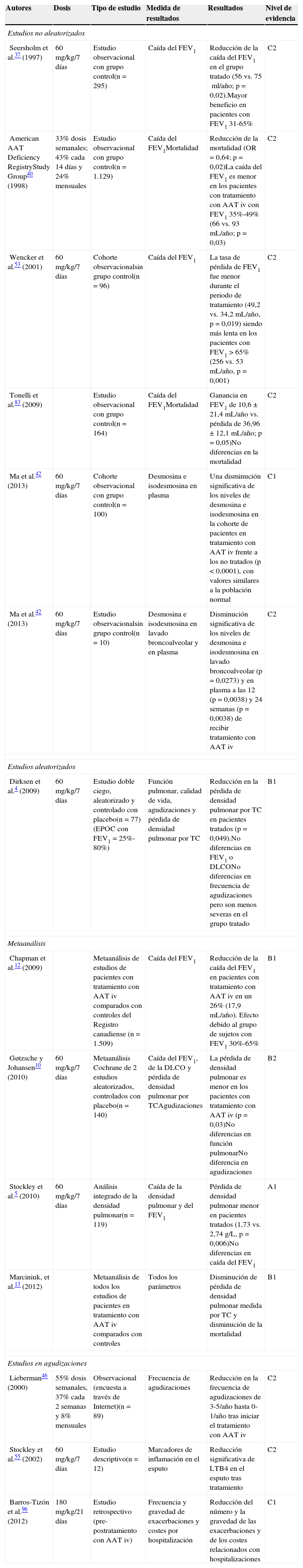
Resumen de los estudios que evalúan el tratamiento con AAT iv
| Autores | Dosis | Tipo de estudio | Medida de resultados | Resultados | Nivel de evidencia |
|---|---|---|---|---|---|
| Estudios no aleatorizados | |||||
| Seersholm et al.37 (1997) | 60mg/kg/7 días | Estudio observacional con grupo control(n = 295) | Caída del FEV1 | Reducción de la caída del FEV1 en el grupo tratado (56 vs. 75ml/año; p = 0,02).Mayor beneficio en pacientes con FEV1 31-65% | C2 |
| American AAT Deficiency RegistryStudy Group40 (1998) | 33% dosis semanales; 43% cada 14 días y 24% mensuales | Estudio observacional con grupo control(n = 1.129) | Caída del FEV1Mortalidad | Reducción de la mortalidad (OR = 0,64; p = 0,02)La caída del FEV1 es menor en los pacientes con tratamiento con AAT iv con FEV1 35%-49% (66 vs. 93mL/año; p = 0,03) | C2 |
| Wencker et al.53 (2001) | 60mg/kg/7 días | Cohorte observacionalsin grupo control(n = 96) | Caída del FEV1 | La tasa de pérdida de FEV1 fue menor durante el periodo de tratamiento (49,2 vs. 34,2mL/año, p = 0,019) siendo más lenta en los pacientes con FEV1 > 65% (256 vs. 53mL/año, p = 0,001) | C2 |
| Tonelli et al.83 (2009) | Estudio observacional con grupo control(n = 164) | Caída del FEV1Mortalidad | Ganancia en FEV1 de 10,6±21,4mL/año vs. pérdida de 36,96±12,1mL/año; p = 0,05)No diferencias en la mortalidad | C2 | |
| Ma et al.42 (2013) | 60mg/kg/7 días | Cohorte observacional con grupo control(n = 100) | Desmosina e isodesmosina en plasma | Una disminución significativa de los niveles de desmosina e isodesmosina en la cohorte de pacientes en tratamiento con AAT iv frente a los no tratados (p < 0,0001), con valores similares a la población normal | C1 |
| Ma et al.42 (2013) | 60mg/kg/7 días | Estudio observacionalsin grupo control(n = 10) | Desmosina e isodesmosina en lavado broncoalveolar y en plasma | Disminución significativa de los niveles de desmosina e isodesmosina en lavado broncoalveolar (p = 0,0273) y en plasma a las 12 (p = 0,0038) y 24 semanas (p = 0,0038) de recibir tratamiento con AAT iv | C2 |
| Estudios aleatorizados | |||||
| Dirksen et al.4 (2009) | 60mg/kg/7 días | Estudio doble ciego, aleatorizado y controlado con placebo(n=77) (EPOC con FEV1 = 25%-80%) | Función pulmonar, calidad de vida, agudizaciones y pérdida de densidad pulmonar por TC | Reducción en la pérdida de densidad pulmonar por TC en pacientes tratados (p = 0,049).No diferencias en FEV1 o DLCONo diferencias en frecuencia de agudizaciones pero son menos severas en el grupo tratado | B1 |
| Metaanálisis | |||||
| Chapman et al.12 (2009) | Metaanálisis de estudios de pacientes con tratamiento con AAT iv comparados con controles del Registro canadiense (n = 1.509) | Caída del FEV1 | Reducción de la caída del FEV1 en pacientes con tratamiento con AAT iv en un 26% (17,9mL/año). Efecto debido al grupo de sujetos con FEV1 30%-65% | B1 | |
| Gøtzsche y Johansen10 (2010) | 60mg/kg/7 días | Metaanálisis Cochrane de 2 estudios aleatorizados, controlados con placebo(n = 140) | Caída del FEV1, de la DLCO y pérdida de densidad pulmonar por TCAgudizaciones | La pérdida de densidad pulmonar es menor en los pacientes con tratamiento con AAT iv (p = 0,03)No diferencias en función pulmonarNo diferencia en agudizaciones | B2 |
| Stockley et al.5 (2010) | 60mg/kg/7 días | Análisis integrado de la densidad pulmonar(n = 119) | Caída de la densidad pulmonar y del FEV1 | Pérdida de densidad pulmonar menor en pacientes tratados (1,73 vs. 2,74g/L, p = 0,006)No diferencias en caída del FEV1 | A1 |
| Marciniuk, et al.13 (2012) | Metaanálisis de todos los estudios de pacientes en tratamiento con AAT iv comparados con controles | Todos los parámetros | Disminución de pérdida de densidad pulmonar medida por TC y disminución de la mortalidad | B1 | |
| Estudios en agudizaciones | |||||
| Lieberman46 (2000) | 55% dosis semanales, 37% cada 2 semanas y 8% mensuales | Observacional (encuesta a través de Internet)(n=89) | Frecuencia de agudizaciones | Reducción en la frecuencia de agudizaciones de 3-5/año hasta 0-1/año tras iniciar el tratamiento con AAT iv | C2 |
| Stockley et al.55 (2002) | 60mg/kg/7 días | Estudio descriptivo(n=12) | Marcadores de inflamación en el esputo | Reducción significativa de LTB4 en el esputo tras tratamiento | C2 |
| Barros-Tizón et al.96 (2012) | 180mg/kg/21 días | Estudio retrospectivo (pre-postratamiento con AAT iv) | Frecuencia y gravedad de exacerbaciones y costes por hospitalización | Reducción del número y la gravedad de las exacerbaciones y de los costes relacionados con hospitalizaciones | C1 |
AAT: alfa-1-antitripsina; iv: intravenosa; DAAT: déficit de alfa-1-antitripsina; DLCO: capacidad de difusión de monóxido de carbono; FEV1: volumen espiratorio forzado en el primer segundo; LTB4: leucotrieno B4; TC: tomografía computarizada.
El grupo de trabajo, sobre la base del nivel de pruebas aportado por 13 estudios específicos sobre el tratamiento con AAT iv4,5,10,12,13,37,40,42,46,53,55,83,96, considera que este tratamiento está indicado en pacientes con DAAT grave (definido como una concentración de AAT ≤ 50mg/dL, medida por nefelometría), no fumadores o exfumadores, con diagnóstico de EPOC y función pulmonar deteriorada (FEV1 < 80% del valor predicho) en los que se documenta pérdida de función pulmonar o progresión del enfisema, a pesar del tratamiento farmacológico y no farmacológico optimizado de la EPOC. El tratamiento con AAT iv no está indicado en heterocigotos Pi*MZ ni en la mayoría de los Pi*SZ, salvo en casos infrecuentes de heterocigotos SZ que presenten concentraciones séricas iguales o inferiores a 50mg/dL y que cumplan el resto de los criterios recogidos en las tablas 3 y 4 (recomendación consistente con calidad de evidencia moderada y acorde a la normativa SEPAR)3. En la enfermedad hepática por DAAT no está indicado el tratamiento con AAT por vía iv.
Criterios del REDAAT para tratamiento con AAT iva
| 1. Mayores de 18 años |
| 2. DAAT demostrado por concentraciones séricas ≤ 50 mg/dL |
| 3. No fumadores o exfumadores al menos durante los últimos 6 meses |
| 4. Enfisema pulmonar demostrado por pruebas de función pulmonar y/o TCAR de tórax |
| 5. EPOC con FEV1< 80% del predichob, que reciben tratamiento farmacológico y no farmacológico óptimo |
| 6. Que no presenten un déficit de inmunoglobulina A |
| 7. Que estén dispuestos a recibir regularmente el tratamiento en hospital de día |
AAT: alfa-1 antitripsina; iv: intravenosa; DAAT: déficit de alfa-1 antitripsina; EPOC: enfermedad pulmonar obstructiva crónica; FEV1: volumen espiratorio forzado espirado en el primer segundo; REDAAT: Registro Español de Pacientes con Déficit de Alfa-1 Antitripsina; TCAR: tomografía computarizada de alta resolución.
Procedimiento a seguir de forma previa al inicio del tratamiento con AAT iv
| Consentimiento informadoa |
| Pruebas complementarias |
| Determinación de inmunoglobulinas séricas |
| Analítica hepática completa |
| Investigar virus de hepatitis-B y virus de la inmunodeficiencia humana |
| Pruebas de función pulmonar: espirometría, volúmenes pulmonares y capacidad de difusión del monóxido de carbono |
| Gasometría arterial: si la saturación periférica de oxígeno es inferior al 92% |
| TCAR de tórax |
| Vacunación frente a virus de la hepatitis B |
AAT: alfa-1 antitripsina; iv: intravenosa; TCAR: tomografía computarizada de alta resolución.
Disponibles en la web del Registro Español del Déficit de Alfa-1 Antitripsina (http://www.redaat.es/presentacion.php) y en el Portal de Salud de la Consejería de la Junta de Andalucía en su área de Consentimientos Informados de Neumología (http://www.juntadeandalucia.es/salud/sites/csalud/contenidos/Informacion_General/p_3_p_11_procedimiento_consentimiento_informado/neumologia?perfil=org).
En relación con los resultados de 3 estudios6,33,48 sobre prevalencia del DAAT en pacientes con bronquiectasias y un metaanálisis13, el grupo de trabajo del REDAAT no recomienda la determinación de concentraciones de AAT de forma habitual en los pacientes con bronquiectasias. Esta práctica debe ser individualizada (recomendación consistente con calidad evidencia baja).
Teniendo en cuenta los resultados de 5 estudios sobre prevalencia del DAAT en asmáticos7,49,54,56,70 y un metaanálisis13, los autores no recomiendan la determinación de las concentraciones de AAT de forma habitual en estos pacientes. Esta práctica debe ser individualizada (recomendación consistente con calidad evidencia baja).
DiscusiónLos resultados expuestos avalan la recomendación de descartar el DAAT en todos los pacientes con EPOC. Esta recomendación ya fue propuesta por la Organización Mundial de la Salud en 199735 y fue posteriormente recogida por diversas normativas, incluidas las de la Sociedad Americana de Tórax (ATS), la europea (ERS) y la española (SEPAR)1,3. Además, aunque no se dispone de estudios suficientes para establecer con precisión un grado de recomendación, los autores aconsejan descartar también el DAAT en los familiares consanguíneos del caso-índice, incluso si están asintomáticos, por la alta probabilidad de que algunos sean portadores de mutaciones graves y puedan beneficiarse del consejo genético y medidas preventivas (la más importante, evitar la inhalación de humo de tabaco y de otros polutantes)1,3.
Con respecto a otras enfermedades obstructivas de las vías aéreas, el grupo de trabajo del REDAAT coincide con otros autores13 en no recomendar la determinación de niveles de AAT para descartar déficit grave de forma habitual en pacientes con bronquiectasias ni en asmáticos, dejando al criterio de los médicos responsables la decisión de individualizar esta prueba en casos concretos, por ejemplo, en pacientes que asocien lesiones de enfisema a las patologías antes citadas.
La determinación cuantitativa de la AAT en suero constituye la base del diagnóstico del DAAT y el método más comúnmente utilizado para realizarla es la nefelometría. Cuando la concentración de AAT es inferior al intervalo normal, es necesario completar el estudio mediante la identificación del fenotipo (variantes proteicas o alélicas). La combinación de ambas técnicas es suficiente para aclarar la mayor parte de los casos de DAAT. El método más utilizado para la identificación de variantes alélicas es el isoelectroenfoque, que puede caracterizar hasta 30 variantes deficitarias de AAT.
Dado que a cada fenotipo le corresponde un rango de valores de AAT, en los casos en los que no haya concordancia entre las concentraciones de AAT y el fenotipo se debe sospechar la presencia de alelos nulos o variantes deficitarias poco frecuentes y, en consecuencia, se debe realizar la determinación del genotipo95. La secuenciación del gen de la AAT mediante la reacción en cadena de la polimerasa a tiempo real (PCR) es el método de referencia para el esclarecimiento de estos casos discordantes (fig. 1)3,95.
. Tomado de Vidal et al.3.")
Algoritmo diagnóstico del déficit de AAT (DAAT).
Tomado de Vidal et al.3.
Las muestras en sangre desecada usadas en programas de cribado informan de la presencia o ausencia de los alelos estudiados, pero no permiten excluir la presencia de otros alelos deficitarios, por lo que será necesario realizar un estudio del genotipo, en los casos en los que no haya concordancia entre la concentración de AAT y el fenotipo. Actualmente, se puede realizar la secuenciación mediante PCR para el estudio del genotipo en las muestras de sangre desecada. Estos métodos son fiables pero es importante que cada laboratorio informe del método utilizado y de sus posibles limitaciones.
En cuanto al tratamiento, el grupo de trabajo del REDAAT considera que:
- –
El tratamiento de los individuos con EPOC asociado a DAAT grave debe incluir el tratamiento farmacológico y no farmacológico recomendado en las normativas de la EPOC111.
- –
Existen suficientes evidencias disponibles, aunque de calidad moderada 4,5,10,12,13,37,40,42,46,53,55,83,96, para recomendar el tratamiento con AAT iv a los individuos con EPOC asociada a DAAT grave (concentraciones séricas de AAT ≤ 50mg/dL), no fumadores o exfumadores, cuyo FEV1 sea inferior al 80% del predicho y presenten pérdida de función pulmonar o progresión del enfisema, a pesar del tratamiento estándar de la EPOC.
- –
La terapia con infusión iv periódica de AAT es el único tratamiento específico que existe para frenar la progresión del enfisema asociado al DAAT. Su eficacia ha sido demostrada en estudios aleatorizados, doble ciego, controlados con placebo, con el análisis de la caída de la densidad pulmonar como parámetro principal de medida.
En la tabla 3 se especifican los requisitos que el REDAAT considera necesarios para aplicar el tratamiento con AAT iv, en la tabla 4 se pormenoriza el procedimiento que se debe seguir antes de iniciar el tratamiento y en la tabla 2 se recogen los principales estudios sobre su eficacia o efectividad en pacientes con EPOC y DAAT grave, y su grado de evidencia.
La eficacia del tratamiento con AAT iv se define sobre la base de los criterios bioquímicos y clínicos. La eficacia bioquímica ha sido demostrada ya que, su administración iv eleva los valores séricos por encima de los considerados protectores, aumenta su concentración en el fluido alveolar y neutraliza la elastasa neutrofílica18,19. Se admite que el valor sérico de AAT que protege al pulmón frente a la elastasa libre del neutrófilo debe ser igual o superior a 50mg/dL (si se determina por nefelometría), a 80mg/dL (si la medición se hace por inmunodifusión radial) o a 11μM/L (si se utiliza el estándar NHLBI del Registro EE. UU.). Este criterio de laboratorio, ampliamente difundido y aplicado, se basa en los trabajos de Wewers et al. (año 1987)18 y Turino et al. (año 1996)34. El estudio de Wewers et al.18 demuestra la eficacia bioquímica de la infusión iv de AAT (nivel de evidencia moderado) pero es inadecuado para establecer un valor de corte protector. El trabajo de Turino et al.34 describe las características clínicas de 56 sujetos SZ, de los que el 52% estaba recibiendo o había recibido tratamiento con AAT iv, y ha sido utilizado para justificar los criterios bioquímicos de selección de pacientes para tratamiento con AAT por vía iv. Sin embargo, el nivel de evidencia científica del estudio es muy bajo y sus hallazgos no pueden ser extrapolados al resto de los SZ. Similares argumentos son aplicables a otro trabajo descriptivo en 25 sujetos SZ17.
Diferentes estudios han demostrado la eficacia clínica del tratamiento con AAT iv4,10,12,13,37,40,42,46,53,55,83,96. El más importante es el estudio de Dirksen et al.4. Se trata de un ensayo aleatorizado, doble ciego, controlado con placebo, cuya variable primaria era la pérdida de densidad pulmonar medida por TC, que demostró una pérdida anual de densidad pulmonar significativamente menor en los sujetos que recibieron tratamiento con AAT iv que en los que no lo recibieron. No hubo diferencias en la función pulmonar, las exacerbaciones y la calidad de vida (cuestionario de St George's) entre ambos grupos. Un análisis posterior realizado por Stockley et al.5, con los datos combinados de estos 2 ensayos, confirmó que los sujetos con tratamiento con AAT iv perdían menos tejido pulmonar que los del grupo placebo (p = 0,006).
Un metaanálisis de Chapman et al., realizado sobre 5 estudios y que incluyó a 1.509 pacientes, encontró que el tratamiento con AAT iv reducía significativamente la pérdida anual de FEV1, sobre todo en los pacientes con un FEV1 entre 30 y 65% del predicho12.
En otro metaanálisis, Gøtzsche y Johansen10 concluyeron que el tratamiento con AAT iv no puede ser recomendado sobre la base de su falta de eficacia y su elevado coste. Sin embargo, este análisis ha sido muy criticado por la comunidad científica y asociaciones de pacientes, como la Alpha-One Fundation, por su parcialidad. Basa sus conclusiones en 2 estudios con un total de 140 pacientes y resta importancia a la medición de la pérdida de densidad pulmonar por TC, cuando esta pérdida es un hecho crucial en la historia natural de estos pacientes. Además, excluye los resultados de algunos estudios observacionales que apoyan la eficacia clínica del tratamiento con AAT iv y que han sido la base para su indicación en las guías de la ATS, la ERS y el American College of Physicians, incluido el estudio multicéntrico, prospectivo de cohortes realizado en 1.129 de pacientes con DAAT del Registro Americano. Este estudio demostró, en el subgrupo de pacientes con valores de FEV1 del 35 al 49%, una disminución del 36% en la mortalidad (RR=0,64) y una reducción significativa de la caída del FEV140.
Finalmente, un reciente metaanálisis muy riguroso de la Sociedad Torácica Canadiense13 recomienda la terapia aumentativa en pacientes con EPOC y FEV1 entre 25 y 80%, no fumadores o exfumadores, con DAAT documentado (μ11μmol/L), que están recibiendo un tratamiento farmacológico y no farmacológico óptimo (incluida rehabilitación), por los beneficios que proporciona (menor pérdida de densidad pulmonar, demostrada por densitometría mediante TC, y reducción de la mortalidad).
En conclusión, el déficit grave de AAT es una condición genética rara cuya manifestación clínica principal es el enfisema pulmonar y existe evidencia suficiente para recomendar el tratamiento con AAT iv (tabla 2) en los pacientes que reúnan ciertas condiciones (tabla 3).
El grupo de trabajo opina que son necesarios estudios para conocer mejor los mecanismos que conducen al desarrollo de EPOC en sujetos con DAAT y para determinar, con una evidencia más firme, cuál es el nivel de AAT capaz de proteger al pulmón de la acción elastolítica de la elastasa, en situación de estabilidad y en caso de agudización, así como la dosis necesaria de AAT para alcanzar estos niveles protectores. Finalmente, los autores opinan que es preciso conseguir medios más efectivos de producción y administración de AAT, que sean más coste-efectivos.
FinanciaciónLos autores no han recibido ningún tipo de financiación para la realización de este artículo.
Conflicto de interesesLa Fundación Española de Pulmón (Respira) ha recibido donaciones de laboratorios Grifols para patrocinar actividades del Registro Español de Pacientes con Déficit de Alfa-1 Antitripsina.
Ana Bustamante ha recibido honorarios por impartir conferencias de Grifols, Astra, Boheringer-Ingelheim, Pfizer, Chiesi, Almirall.
Francisco Casas ha recibido honorarios por asesoría científica y/o por impartir conferencias de Almirall, AstraZeneca, Boehringer Ingelheim, Grupo Ferrer, GlaxoSmithKline, Grifols, Laboratorios Esteve, Pfizer, Novartis y Takeda.
José María Hernández ha recibido honorarios de Grifols por asesoría científica y por impartir conferencias.
Lourdes Lázaro ha recibido honorarios de Grifols por impartir conferencias.
Beatriz Lara ha recibido honorarios por impartir conferencias de Boehringer Ingelheim, Pfizer, Grifols y Novartis.
Marc Miravitlles ha recibido honorarios por asesoría científica y/o por impartir conferencias de Almirall, AstraZeneca, Bayer Schering, Boehringer Ingelheim, Grupo Ferrer, GlaxoSmithKline, Grifols, Laboratorios Esteve, Pfizer, Novartis y Nycomed.
María Torres ha recibido honorarios de Grifols por asesoría científica.
El Comité del REDAAT agradece a los Dres. Rafael Vidal, Rosendo Jardí, Juan Carlos Barros-Tizón, Pedro Pablo España y Carlos Escudero su contribución durante años a las actividades del Registro.







