


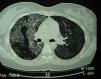

Pulmonary alveolar proteinosis (PAP) is a rare disease characterized by the accumulation of surfactant-like lipoproteinaceous material in the distal air spaces and terminal bronchi, which may lead to impaired gas exchange. This accumulation of surfactant is due to decreased clearance by the alveolar macrophages. Its primary but most common form is currently considered an autoimmune disease. Better knowledge of the causes of PAP has led to the emergence of alternatives to whole lung lavage, although this is still considered the treatment of choice. Most studies are case series, often with limited patient numbers, so the level of evidence is low. Since the severity of presentation and clinical course are variable, not all patients will require treatment. Due to the low level of evidence, some objective criteria based on expert opinion have been arbitrarily proposed in an attempt to define in which patients it is best to initiate treatment.
La proteinosis alveolar pulmonar es una enfermedad rara que se caracteriza por la acumulación del material lipoproteináceo del surfactante en los espacios alveolares y los bronquiolos terminales, lo que puede llegar a producir alteraciones en el intercambio gaseoso. Esta acumulación del surfactante es debida a una disminución en su aclaramiento por parte de los macrófagos alveolares. Su forma primaria, la más frecuente, es considerada actualmente una enfermedad autoinmune. El mayor conocimiento de las causas que la provocan ha conducido a la aparición de tratamientos alternativos al lavado pulmonar total, que sigue siendo considerado de elección. La mayoría de los trabajos están constituidos por series de casos, la mayoría de las veces con pocos pacientes, por lo que el nivel de evidencia es bajo. Dado que la gravedad de su presentación y su curso son variables, no todos los pacientes van a requerir tratamiento. Debido al bajo nivel de evidencia de que se dispone, ya que la mayoría de estudios son series de casos, se han propuesto de manera arbitraria algunos criterios objetivos, basados en la opinión de expertos, que intentan definir en qué pacientes es más adecuado iniciar el tratamiento.
Pulmonary alveolar proteinosis (PAP) is a rare disease that was first described in 1958.1 It is characterized by the accumulation of surfactant-like lipoproteinaceous material in the distal air spaces and terminal bronchi, which may lead to impaired gas exchange2 (Fig. 1). This accumulation of surfactant is due to reduced clearance by the alveolar macrophages, and another feature of the disease is the presence of phospholipoproteic material within the alveolar macrophages. Estimated yearly incidence is 0.2–0.4cases/million, and prevalence is 3.7–6.2persons/year.3 As of 2002, around 410 cases had been described in the literature.4
Classification and Pathogenesis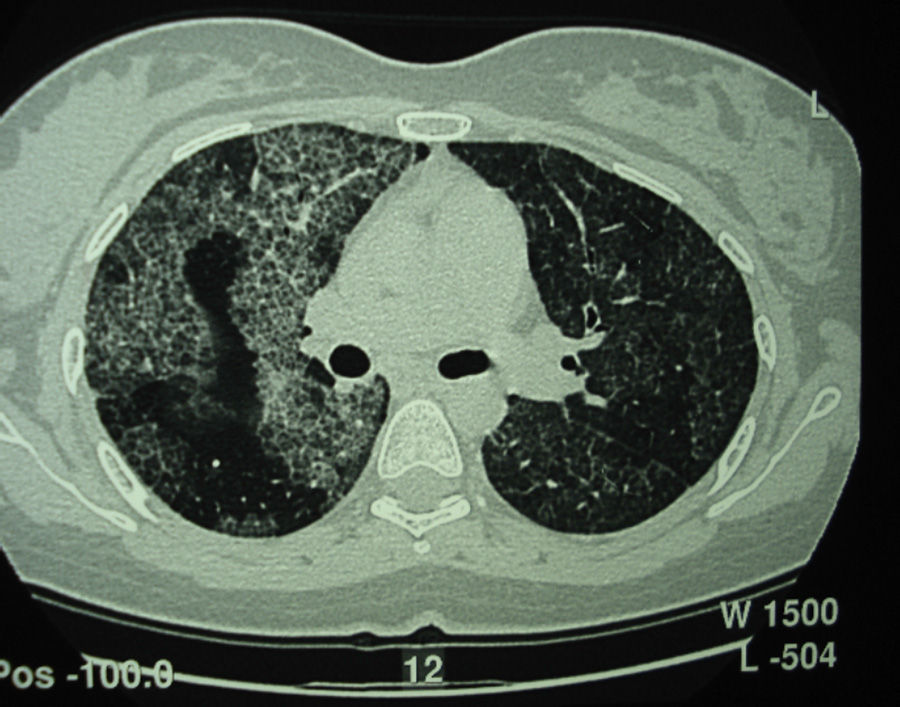
The main elements in the development of PAP are the appearance of defective granulocyte-macrophage colony stimulating factor (GM-CSF) activity or changes in the cell surface GM-CSF receptor and in its signaling mechanisms. GM-CSF is necessary for final alveolar macrophage differentiation and maturing.5 One of the major advances in the understanding of the pathogenic mechanisms of PAP occurred unexpectedly following work carried out in an animal model with GM-CSF knockout mice. Investigators were surprised to find that although these mice showed no hematological manifestations, they developed a pulmonary condition very similar to human PAP. Moreover, lungs recovered once the GM-CSF function was reestablished, irrespective of whether GM-CSF was administered exogenously by inhalation, the non-functioning gene was repaired, or the mice underwent bone marrow transplant.6 Subsequent studies in mice models showed that local GM-CSF deficit produced by inhibiting the GM-CSF gene or by deletion of the GM-CSF/IL-3/IL-5 receptor β-subunit on the cell surface leads to the development of PAP.7
From a clinical point of view, there are 3 main forms of PAP: hereditary or congenital; autoimmune or primary; and secondary. Mechanisms leading to alveolar macrophage dysfunction differ in each of these 3 forms.8
In primary PAP, anti-GM-CSF IgG-type neutralizing antibodies are detected. This form accounts for 90% of all cases of the disease. These antibodies are found in both serum and bronchoalveolar lavage (BAL) fluid, and confirm the autoimmune mechanism involved in primary PAP.9,10 These neutralizing antibodies cause alveolar macrophage dysfunction, affecting catabolism and clearance of surfactant from the distal air spaces.11 In cases of congenital or hereditary PAP, the defective elimination of surfactant is caused by mutations in genes coding for surfactant protein-B (SP-B),12 surfactant protein-C (SP-C),13 or in genes coding for GM-CSF receptor chains (CSF2RA-α14 and CSF2RB-β15), preventing GM-CSF from binding to its membrane receptor.16 Other germline mutations can also cause different diseases, including PAP.17 Secondary PAP, for its part, occurs in several diseases that involve a reduction in the number or function of the alveolar macrophages, such as myelodysplastic syndrome, leukemias or lymphomas.18 PAP may also occur in association with some infections (Nocardia, Pneumocystis jirovecii) or after environmental or occupational exposure to substances such as silica, aluminum, titanium or some fertilizers.19
TreatmentIn this review, we will discuss the treatment of primary or autoimmune PAP, as the treatment of secondary PAP depends mainly on the specific disease with which it is associated. There are currently no firm, universally accepted criteria for starting treatment for PAP, particularly for milder cases, and the decision to initiate specific treatment will depend on the severity of the clinical presentation. This consideration is based on the natural disease course, in which a high percentage of cases show spontaneous remission. In the largest series published to date, patients who were asymptomatic had a high probability of remaining stable or even improving without treatment, and only 8% deteriorated during follow-up. Of those with symptoms, the proportion of patients who remained stable, improved or worsened was 45%, 30%, and 25%, respectively.4 Those with longer disease courses were more likely to deteriorate.
Treatment of choice is still whole lung lavage, although there have been advances in alternative or complementary treatments, such as the administration of GM-CSF, rituximab, plasmapheresis or stem cell transplants. Due to the rarity of this disease—and thus the scant number of patients—most of these treatments are based on the results obtained from case series, rather than randomized clinical trials. Some of them are still in an experimental phase, so the level of evidence in most cases is weak and based solely on expert opinion.
In order to achieve the best risk-benefit ratio when the decision has been taken to start treatment, most authors agree to arbitrarily classify the patients into 3 categories or groups,20 as follows:
- •
Group 1. Asymptomatic patients and/or those with mild gas exchange impairment. These patients do not need immediate treatment and must be monitored periodically for symptoms, lung function and imaging studies.
- •
Group 2. Patients with mild/moderate symptoms (dyspnea on exertion) and oxygenation changes during exercise (not at rest). This group should be managed with oxygen therapy during exercise and more closely monitored for any clinical or functional deterioration that may require more aggressive treatment. The recommendation of oxygen therapy in this group is not supported by clinical trial results; rather it is based on expert opinion with little scientific evidence.
- •
Group 3. This group includes patients with moderate or severe symptoms, who have gas exchange impairment at rest. This group needs specific, more aggressive treatment. The treatment of choice is whole lung lavage, and other therapeutic interventions can be adopted, depending on the results obtained and the progress of the disease.
Due to the scant evidence on which treatment is based and to more accurately define cases of PAP that require more specific treatment, some authors have proposed more objective criteria, based primarily on oxygenation factors. Patients with the following parameters would be candidates for treatment: (a) resting PaO2<65mmHg; (b) alveolar-arterial oxygen tension gradient≥40mmHg, and (c)shunt fraction >10%–12%.21
Whole Lung LavageWhole lung lavage remains the treatment of choice for PAP. It was first described over 40 years ago, and since then the technique has undergone very few changes.22 It needs to be performed under general anesthesia with double-lumen endotracheal intubation. One lung is ventilated and oxygenated via one lumen, while the contralateral lung is washed with saline solution at a temperature of approximately 37°C. Aliquots of 1–1.5L of warmed saline solution are usually needed for each lavage, and between 10 and 15 lavages are needed to treat one whole lung. The procedure generally lasts around 3–4h. Chest percussion during the procedure has been reported to significantly increase the extraction of lipoproteinaceous material.23 Lavage of the other lung can be done on a separate occasion or sequentially in the same session.
Oxygenation and pulmonary mechanics must be monitored during the procedure. The double-lumen tube must be carefully positioned and care must be taken to recover the saline solution that has been instilled, to avoid possible complications caused by poor placement of the endotracheal tube, passage of serum to the ventilated lung, or the development of hydropneumothorax. Other complications include pneumonia, laryngospasm or arrhythmias.24
In 75%–95% of cases, symptoms, oxygenation capacity, and radiological changes improve rapidly after whole lung lavage, generally in the first few days after the procedure.19 This benefit tends to be sustained for a mean period of 15 months, although relapses are common, and occur in up to 45%–70% of cases over the subsequent 3 years. However, whole lung lavage does not need to be repeated in all cases of relapse. The results of the different series vary widely, and repeating whole lung lavage has been reported in a range of between 15% and 70% of cases. It should be emphasized that around 30%–40% of cases will require only one lavage. It is important to note that 5-year survival is greater in patients who have undergone whole lung lavage that in those who have not (94% vs 85%, P<.04).4
Subcutaneous GM-CSFAfter anti-GM-CSF antibodies in the serum and BAL of patients with primary PAP and their role in PAP pathogenesis were discovered, the possibility of exogenous administration of this cytokine was proposed as a potential treatment. No randomized clinical trials have been performed and most published papers are observational studies including small numbers of patients25,26 (Table 1). Seymour et al.27 published the first case of clinical improvement in an adult receiving GM-CSF subcutaneously. This author subsequently treated another 14 patients28 at a dose of 3μg/kg/day for 5 days, increasing to 5μg/kg/day for a period of 12 weeks. One patient discontinued treatment after 13 days due to neutropenia, and 5 discontinued after 6 weeks due to lack of response. Seven patients continued treatment for 12 weeks and one patient was treated for 26 weeks. Treatment was considered effective in 6/14 (43%, 95% CI 18%–71%), with a mean treatment period of 39 weeks. Of these 6 cases, 5 worsened after discontinuation of the therapy, and 4 improved after it was resumed.
Summary of Studies Published Investigating Treatments Other Than Whole Lung Lavage.20
| Author | Intervention | Dose | Duration | Response |
|---|---|---|---|---|
| Seymour et al.28 | GM-CSF sc | 5mg/kg/day | 10–26 weeks | 36% (n=14) |
| Kavuru et al.39 | GM-CSF sc | 250μg/day to 5–9μg/kg/day | 12 weeks | 75% (n=4) |
| Bonfield et al.30 | GM-CSF sc | 250μg/day to 18μg/kg/day | 12–48 weeks | 55% (n=11) |
| Venkateshiah et al.31 | GM-CSF sc | 250μg/day to 5–18μg/kg/day | 12–52 weeks | 48% (n=21) |
| Tazawa et al.32 | GM-CSF inh | 250μg/day alternate weeks | 24 weeks | 100% (n=3) |
| Wylam et al.33 | GM-CSF inh | 250–500μg/day | 12 weeks | 83% (n=12) |
| Borie et al.34 | Rituximab iv | 1g day 0 and day 15 | 15 days | 100% (n=1) |
| Amital et al.35 | Rituximab iv | 375mg/m2 weekly | 4 weeks | 100% (n=1) |
| Kavuru et al.36 | Rituximab iv | 1g day 0 and day 15 | 15 days | 78% (n=9) |
inh: inhaled; iv: intravenously; sc: subcutaneously.
Kavuru et al.29 treated a group of 4 patients for 12 weeks. The dose for the first 4 weeks was 250μg/day, with daily 5μg/kg increments during the next 4 weeks, and finally, 9μg/kg/day for the last 4 weeks. Three of the 4 patients experienced improved lung function, oxygenation and exercise capacity.
In a prospective trial, Bonfield et al.30 included 14 cases of PAP, of whom 11 completed the study. All patients had previously undergone whole lung lavage. The initial dose was 250μg/day, with dose increases every 2 weeks to a maximum of 18μg/kg/day. Total time on treatment was 12–48 weeks. Six patients experienced improved oxygenation, from 69.1±3.4mmHg to 84.0±2.0mmHg. The group that responded to treatment showed reduced levels of anti-GM-CSF antibodies in both serum and BAL.
In a prospective open-label clinical trial in 25 patients,31 12 (57%) of the 21 patients that completed the trial showed significant improvement, and 6 (67%) did not need either whole lung lavage or home oxygen therapy.
Despite the methodological limitations, the range of doses used and the variable duration of treatment, subcutaneous administration of GM-CSF appears to be effective in up to 2/3 of cases. It has a few side effects, including edema and erythema in the injection site and low-grade fever; no changes are generally seen in blood tests. Subcutaneous GM-CSF can be considered an alternative treatment for PAP.
Inhaled GM-CSFTazawa et al.32 reported a series of 50 patients with primary PAP who were given inhaled GM-CSF at a dose of 125μg every 12h for 1 week, followed by 1 rest week, for a total of six 2-week cycles (12 weeks of treatment in all). They were followed up for 52 weeks after completion of the treatment. Of the 35 patients who completed the trial, 24 showed clear improvement in alveolar-arterial oxygen gradient (12.3mmHg, 95% CI 8.4–16.2mmHg, P<.001), with an overall improvement in 62% (in 24/39 patients in the final intent-to-treat analysis). During follow-up, 29 of the 35 patients were clinically stable, and did not require any additional treatment.
Wylam et al.33 carried out a retrospective study in 12 patients with primary PAP who received increasing doses of GM-CSF in aerosol up to a maximum of 500μg/day for a period of 12 weeks. Results were positive, with 11 patients achieving improved symptoms, and 10 achieving significantly improved gas exchange parameters. These results were maintained in the long-term: 8 patients achieved partial remission and 3 achieved complete radiological remission.
Inhaled GM-CSF treatment can be considered effective in 4/5 cases, with few side effects. Doses and treatment duration are variable.
RituximabRituximab is a monoclonal antibody that targets the CD20 antigen receptor on the surface of the B-cells. This target is a 35kD membrane protein that plays an essential role in the cell cycle and B-cell differentiation.34 Administration of rituximab causes a rapid reduction of B-cells in peripheral blood. Several mechanisms of action have been suggested: complement-mediated cytotoxicity, antibody-dependent cell-mediated cytotoxicity, or increased antibody-mediated apoptosis. Rituximab is used in the treatment of lymphoproliferative diseases of the B-cells and in various autoimmune diseases such as systemic lupus erythematosus, rheumatoid arthritis or vasculitis.35 As primary PAP is considered an autoimmune disease involving anti-GM-CSF antibodies, the reduction of B-cells and anti-GM-CSF levels induced by rituximab may be an effective treatment option.36
Borie et al.37 described the effect of rituximab in a PAP patient who refused lung lavage. Two 1g doses were administered 15 days apart, producing a fall in serum B-cells and anti-GM-CSF levels. After 6 months of treatment, the alveolar-arterial gradient had improved but DLCO and HRCT results were unchanged. Amital et al.38 described the effect of rituximab in a PAP patient who showed no improvement after whole lung lavage and subsequent administration of subcutaneous GM-CSF. She received a weekly dose of 375mg/m2 for 4 weeks, showing improved gas exchange, DLCO and radiological findings.
The largest study to date is an open-label clinical trial with rituximab in 10 PAP patients.39 Two 1g doses were administered on day 0 and day 15. Alveolar-arterial oxygen gradient and PaO2 improved in 7 of 9 patients who completed the trial. Improvement was sustained at 3 and 6 months after treatment completion. Improved findings on HRCT and lung function tests were also observed.
Rituximab treatment is not free of side effects; some of which are serious. Although the results of trials have been promising, its use in PAP is still is an experimental phase.
Several clinical trials evaluating both rituximab and GM-CSF in PAP are currently ongoing: for more information, see http://apps.who.int/trialsearch/.
PlasmapheresisPlasmapheresis for reducing anti-GM-CSF levels in serum has been attempted in several isolated cases in which other treatments, such as lung lavage, anti-GM-CSF or rituximab, were ineffective. Results have been varied, and its efficacy as a treatment for PAP has not yet been established.40,41
Lung TransplantationLung transplantation has been performed in cases in which other treatments were ineffective and the disease progressed. Isolated cases of lung transplantation in adults have been reported, while in a series of 270 transplantations performed in 190 children between 1990 and 2002, the indication in 12 (6.3%) was PAP.42 This is an option for severe, treatment-refractory cases. Recurrent PAP in the transplanted lung at 3 years has been reported,43 but the incidence of recurrence is unknown. If new treatments can be developed for PAP, transplantation may be confined to very exceptional cases.
CorticosteroidsCorticosteroids are not indicated in the treatment of PAP, and indeed, they may worsen prognosis, since they can interfere with surfactant metabolism44 and alter the immune response.4 In early papers published on PAP, fatalities associated with the development of norcardiosis, cryptococcosis or mucormycosis were reported in patients treated with corticosteroids.45
Allogenic Hematopoietic Stem Cell TransplantationPAP was reversed in a mouse model after hematopoietic stem cell transplantation, and reports of good response to this treatment in secondary or hereditary PAP have been published.5 However, it is a treatment that is still under investigation and for which very little evidence is available. A new line of research that would avoid prior ablative treatment may be alveolar macrophage transplantation.46 This approach has shown promising results in mouse models and may become a treatment option in the future.47
Finally, these patients may have bacterial infection who need appropriate antibiotic treatment. Defective macrophage activity in the lungs heightens susceptibility to infection from Nocardia or Pneumocystis jirovecii, for example. For this reason, some authors recommend prophylaxis with trimethoprim/sulfamethoxazole until complete disease remission.48
ConclusionThe treatment of choice for PAP is still whole lung lavage, but it is not required in all patients. Better understanding of the pathogenic mechanisms of this disease has led to more therapeutic options (Fig. 2). GM-CSF, both inhaled and subcutaneous, has been used successfully in primary or autoimmune PAP, but the optimal dose, treatment duration and route of administration have not yet been definitively established. Other promising treatments are emerging, but they are still in the research phase. As this is a very rare disease, and some patients remit spontaneously, it is difficult to establish a high level of evidence for these treatments. Perhaps combined therapies will be used for more severe cases in the future.
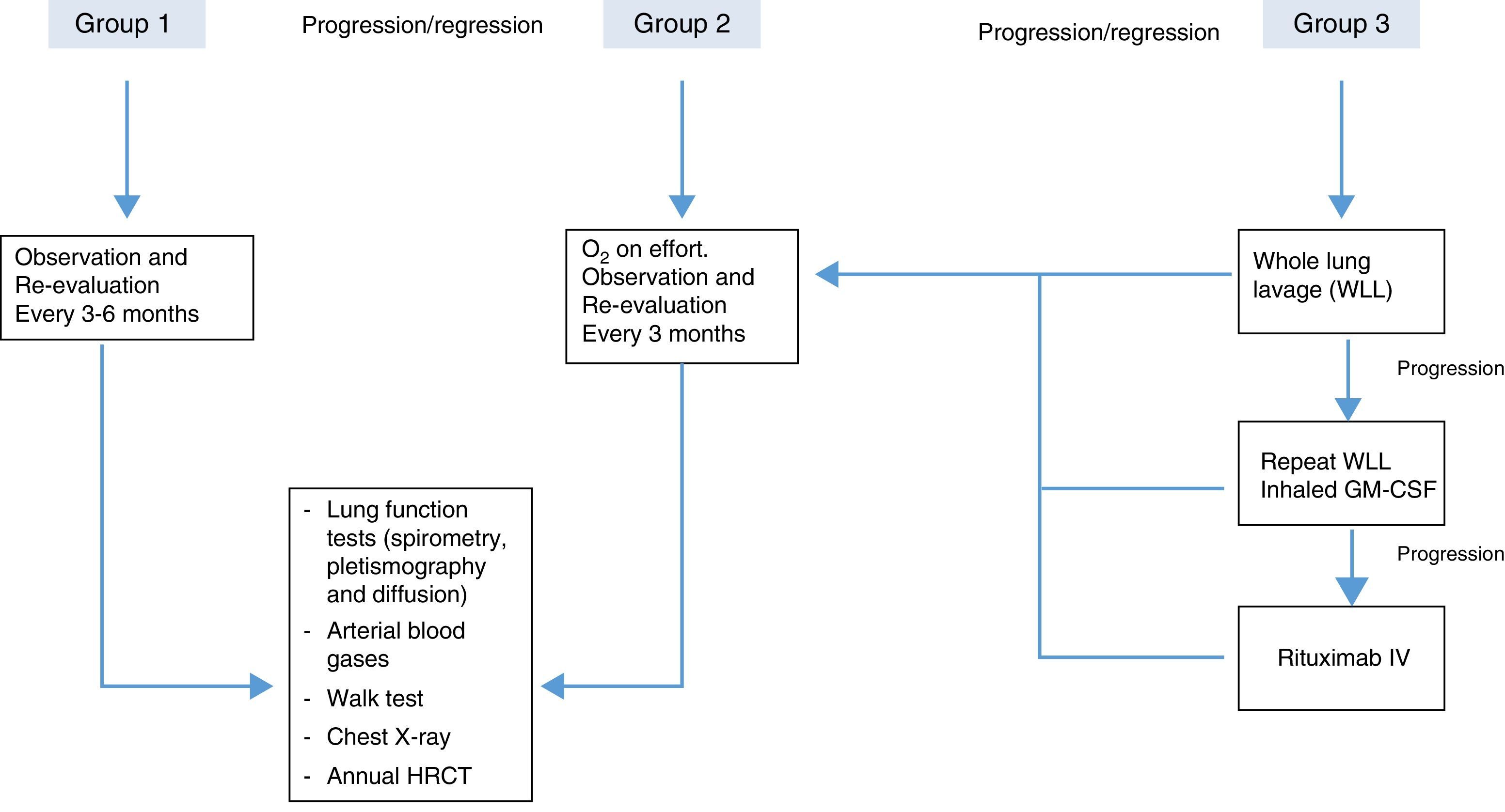
Suggested PAP-treatment algorithm, according to severity, based on the opinions of different authors.20
The authors declare that they have no conflict of interests.
Please cite this article as: Rodríguez Portal JA. Tratamiento de la proteinosis alveolar primaria del adulto. Arch Bronconeumol. 2015;51:344–349.







