




La proteinosis alveolar pulmonar es una enfermedad rara que se caracteriza por la acumulación del material lipoproteináceo del surfactante en los espacios alveolares y los bronquiolos terminales, lo que puede llegar a producir alteraciones en el intercambio gaseoso. Esta acumulación del surfactante es debida a una disminución en su aclaramiento por parte de los macrófagos alveolares. Su forma primaria, la más frecuente, es considerada actualmente una enfermedad autoinmune. El mayor conocimiento de las causas que la provocan ha conducido a la aparición de tratamientos alternativos al lavado pulmonar total, que sigue siendo considerado de elección. La mayoría de los trabajos están constituidos por series de casos, la mayoría de las veces con pocos pacientes, por lo que el nivel de evidencia es bajo. Dado que la gravedad de su presentación y su curso son variables, no todos los pacientes van a requerir tratamiento. Debido al bajo nivel de evidencia de que se dispone, ya que la mayoría de estudios son series de casos, se han propuesto de manera arbitraria algunos criterios objetivos, basados en la opinión de expertos, que intentan definir en qué pacientes es más adecuado iniciar el tratamiento.
Pulmonary alveolar proteinosis (PAP) is a rare disease characterized by the accumulation of surfactant-like lipoproteinaceous material in the distal air spaces and terminal bronchi, which may lead to impaired gas exchange. This accumulation of surfactant is due to decreased clearance by the alveolar macrophages. Its primary, most common form, is currently considered an autoimmune disease. Better knowledge of the causes of PAP have led to the emergence of alternatives to whole lung lavage, although this is still considered the treatment of choice. Most studies are case series, often with limited patient numbers, so the level of evidence is low. Since the severity of presentation and clinical course are variable, not all patients will require treatment. Due to the low level of evidence, some objective criteria based on expert opinion have been arbitrarily proposed in an attempt to define in which patients it is best to initiate treatment.
La proteinosis alveolar pulmonar (PAP) es una enfermedad rara que fue descrita por primera vez en el año 19581. Se caracteriza por la acumulación del material lipoproteináceo del surfactante en los espacios alveolares y bronquiolos terminales, lo que puede llegar a producir alteraciones en el intercambio gaseoso2 (fig. 1). Esta acumulación del surfactante es debida a una disminución en su aclaramiento por parte de los macrófagos alveolares, siendo característica la presencia de macrófagos alveolares con inclusiones de material fosfolipoproteico. Su incidencia estimada es de 0,2-0,4 casos por millón de personas/año, con una prevalencia de 3,7-6,2 personas/año3. Hasta el año 2002 se habían descrito en la literatura unos 410 casos4.
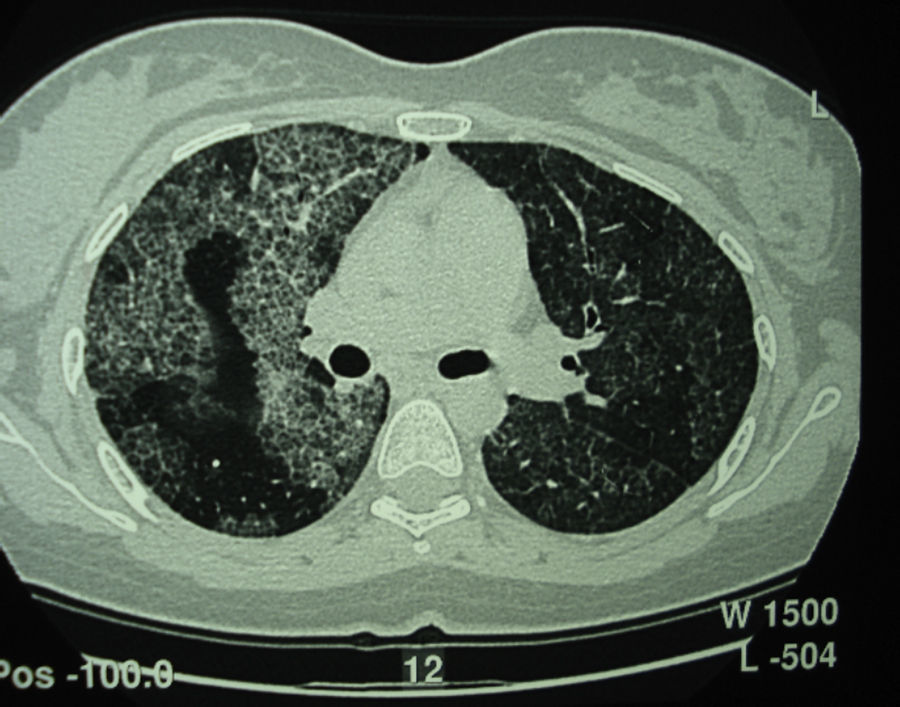
Tomografía computarizada de alta resolución (TACAR) de un pacientes con proteinosis alveolar pulmonar (PAP) que muestra las características clásicas de los septos interlobulillares engrosados contra un fondo de opacidades en vidrio esmerilado, dando la típica una apariencia de crazy paving.
Los elementos principales para que se desarrolle la PAP son la aparición de deficiencias en la actividad del factor estimulante de colonias de granulocitos-macrófagos (GM-CSF) o bien alteraciones en el receptor del GM-CSF en la superficie celular y en sus mecanismos de señalización. El GM-CSF es necesario para que se produzca la diferenciación final y la maduración del macrófago alveolar5. Uno de los mayores avances en la comprensión de los mecanismos patogénicos de la PAP se produjo de una manera inesperada gracias a la investigación animal en ratones Knockout para el gen que codifica el GM-CSF. Estos ratones sorprendentemente no tenían más manifestaciones hematológicas pero desarrollaban un cuadro pulmonar superponible a la PAP humana. Además, se demostró que los pulmones se recuperaban una vez restablecida la función del GM-CSF tanto si era administrado de manera exógena por vía inhalatoria, como si se reparaba el gen no funcionante o si los ratones se sometían a un trasplante de médula ósea6. Estudios posteriores en modelos murinos demostraron que el déficit local de GM-CSF que se produce por la supresión del gen de GM-CSF o bien por la desaparición de la subunidad-β del receptor GM-CSF/IL-3/IL-5 en la superficie celular provocaba la aparición de PAP7.
Desde el punto de vista clínico, hay 3 formas principales de presentación de la PAP (hereditaria o congénita, autoinmune o primaria y secundaria). Los mecanismos que conducen a la disfunción de los macrófagos alveolares difieren en cada una de ellas8.
En la PAP primaria se detectan anticuerpos neutralizantes de la clase IgG anti GM-CSF. Supone el 90% de todos los casos de PAP. Estos anticuerpos se encuentran presentes tanto en el suero como en el líquido de lavado broncoalveolar (BAL) y confirman el mecanismo autoinmune en la patogenia de la PAP primaria9,10. Estos anticuerpos neutralizantes van a ocasionar defectos en la función del macrófago alveolar, alterando el catabolismo y el aclaramiento del surfactante del espacio alveolar11. En los casos de PAP congénita o hereditaria, el defecto en la eliminación del surfactante es debido a que se producen mutaciones en los genes que codifican la proteína del surfactanteB (SP-B)12, la del surfactanteC (SP-C)13, o en los genes que codifican las cadenas del receptor del GM-CSF (CSF2RA-α14 y CSF2RB-β15), impidiendo la unión del GM-CSF a su receptor de membrana16. También existen algunas mutaciones en genes de células germinales que van a producir varias enfermedades, entre ellas la PAP17. Finalmente, los casos de PAP secundaria se producen en varias enfermedades que provocan un descenso en el número o en la función del macrófago alveolar, como en el síndrome mielodisplásico, leucemias o linfomas18. También pueden aparecer casos de PAP asociada a algunas infecciones (Nocardia, Pneumocystis jirovecii) o tras exposiciones medioambientales y ocupacionales a sustancias como sílice, aluminio, titanio o algunos fertilizantes19.
TratamientoVamos a referirnos al tratamiento de la PAP primaria o autoinmune. En los casos de PAP secundaria el tratamiento va a depender de cada enfermedad específica con la que se asocie. En la actualidad no hay unos criterios firmes y unánimemente aceptados para iniciar algún tratamiento en la PAP, especialmente en los casos leves. La decisión de instaurar un tratamiento específico en la PAP va a depender de la gravedad en la presentación clínica. Esta consideración está basada en la observación de la historia natural de la enfermedad, que refleja un alto porcentaje de casos en los que se produce una remisión espontánea. En la serie publicada con mayor número de casos se comprobó que los enfermos que estaban asintomáticos era muy probable que se mantuvieran estables o incluso mejoraran sin tratamiento, y solo un 8% empeoraban durante el seguimiento. Entre aquellos que tenían síntomas, la proporción de casos que se mantenía estable, mejoraba o empeoraba era del 45, del 30 y del 25%, respectivamente4. En aquellos casos en los que había un mayor tiempo de evolución de la enfermedad, era más probable que hubiera una progresión de los síntomas.
El tratamiento de elección sigue siendo la realización de un lavado pulmonar total, si bien se han ido desarrollando tratamientos alternativos o complementarios, como la administración de GM-CSF, el rituximab, la plasmaféresis o el trasplante de progenitores hematopoyéticos. Debido a la rareza de esta enfermedad —y por tanto el escaso número de pacientes—, la mayoría de estos tratamientos están basados en resultados obtenidos de series de casos, no en ensayos aleatorizados, y algunos de ellos siguen estando en fase de investigación, por lo que el nivel de evidencia en la mayoría de los casos es débil, basada únicamente en la opinión de expertos.
Con el objeto de obtener la mejor relación riesgo/beneficio cuando se decide instaurar algún tratamiento en la PAP, la mayoría de autores están de acuerdo en clasificar de forma arbitraria a los pacientes en 3 categorías o grupos20:
- •
Grupo 1. Aquellos que están asintomáticos y/o presentan mínimas alteraciones en el intercambio gaseoso. Estos pacientes no requieren tratamiento inmediato y deberían ser observados periódicamente con control de síntomas, función pulmonar y técnicas de imagen.
- •
Grupo 2. Los que tienen síntomas leves/moderados (disnea de esfuerzo) y alteraciones en la oxigenación durante el ejercicio (no en reposo). En este grupo debe instaurarse oxigenoterapia al esfuerzo y control más estrecho para identificar algún deterioro clínico o funcional que requiera instaurar tratamientos más agresivos. El tratamiento con oxigenoterapia en este grupo no está basado en ningún ensayo clínico, siendo una recomendación de expertos con poca evidencia científica.
- •
Grupo 3. En este grupo se encuadran los pacientes con síntomas moderados o graves, en los que hay alteraciones en el intercambio gaseoso en reposo. Este grupo es el que requiere tratamiento específico y más agresivo. El tratamiento de elección es el lavado pulmonar total, adoptando otras medidas terapéuticas en función de los resultados obtenidos y la progresión de la enfermedad.
Debido a la escasa evidencia en la que se basa el tratamiento y con el objetivo de ser más preciso al definir aquellos casos de PAP que requieren un tratamiento más específico, algunos autores proponen criterios más objetivos, basados fundamentalmente en las alteraciones que se producen en la oxigenación, de manera que serían candidatos a tratamiento aquellos casos que presentaran: a)una PaO2 en reposo <65mmHg; b)una diferencia alveoloarterial de O2≥40mmHg, y c)presencia de un shunt >10-12%21.
Lavado pulmonar totalSigue siendo la técnica de elección para el tratamiento de la PAP. Fue descrita hace más de 40años, y desde entonces ha experimentado muy pocas modificaciones22. Para su realización es necesaria anestesia general y realizar una intubación con un tubo endotraqueal de doble luz. A través de una luz se procede a la ventilación y a la oxigenación de un pulmón, mientras el pulmón contralateral es lavado con suero salino a una temperatura aproximada de 37°C. Generalmente se precisan alícuotas de 1 a 1,5l de suero salino templado en cada lavado, y suelen necesitarse entre 10 y 15 lavados para conseguir lavar todo un pulmón. El procedimiento suele durar en total unas 3-4h. Se ha descrito que la percusión torácica durante el procedimiento del lavado pulmonar mejora significativamente la extracción del material lipoproteináceo23. El lavado del pulmón contralateral puede hacerse en un segundo tiempo o de manera secuencial en una misma sesión.
Durante el procedimiento es preciso monitorizar la oxigenación y la mecánica pulmonar. Hay que asegurar la correcta posición del tubo de doble luz y comprobar que se consigue la recuperación del suero salino que se ha instilado para evitar la aparición de posibles complicaciones, que suelen producirse por la mala posición del tubo endotraqueal, el paso de suero al pulmón ventilado o la aparición de hidroneumotórax. Otras complicaciones descritas son la aparición de neumonías, laringoespasmo o arritmias24.
Tras el lavado pulmonar total se produce una rápida mejoría de los síntomas, de la capacidad de oxigenación y de las alteraciones radiológicas en un 75-95% de los casos. La mejoría se produce a lo largo de los primeros días tras la realización del procedimiento19. Este beneficio suele mantenerse durante un periodo medio de 15meses, si bien las recaídas son frecuentes, ocurriendo hasta en un 45-70% de los casos a lo largo de los 3años siguientes. Sin embargo, no es preciso repetir el lavado pulmonar total en todos los casos en los que se produce una recaída. Los resultados de las distintas series son muy variables, y el rango en los que se ha repetido el lavado pulmonar total tras una recaída oscila entre el 15 y el 70% de los casos. Hay que resaltar que en el 30-40% de los casos se va a precisar un único lavado. Es importante señalar que la supervivencia a los 5años es superior en el grupo de pacientes que han sido tratados con lavado pulmonar total con respecto a los no tratados (94% frente a 85%, p<0,04)4.
GM-CSF subcutáneoTras el descubrimiento de los anticuerpos antiGM-CSF en el suero y en BAL en los casos de PAP primaria y su papel en la patogenia, se ha ido planteado la posibilidad de la administración de esta citoquina por vía exógena como una posible terapia. No existen ensayos clínicos aleatorizados, y la mayoría de los trabajos publicados son estudios observacionales en los que se incluyen un escaso número de casos25,26 (tabla 1). Seymour et al.27 publicaron el primer caso de un adulto tratado con GM-CSF por vía subcutánea en el que se comprobó una mejoría clínica. Posteriormente, este mismo autor trató a otros 14 pacientes28 administrando una dosis de 3μg/kg/día durante 5días, aumentando a 5μg/kg/día durante un periodo de 12semanas. Uno de ellos abandonó el tratamiento a los 13días por la aparición de neutropenia, y 5 lo suspendieron a las 6semanas por falta de respuesta. En 7 casos el tratamiento se prolongó durante 12semanas, y un caso fue tratado durante 26 semanas. El tratamiento fue considerado eficaz en 6/14 (43%, IC95% 18-71%), con un tiempo medio de tratamiento de 39semanas. De estos 6 casos, en 5 se produjo un deterioro tras la suspensión de la terapia, obteniéndose mejoría en 4 de ellos tras la reintroducción del tratamiento.
Resumen de los diferentes trabajos publicados con tratamientos alternativos al LPT.(ref 20)
| Autor | Intervención | Dosis | Duración | Respuesta |
|---|---|---|---|---|
| Seymour et al.28 | GM-CSF sc | 5mg/kg/día | 10-26 semanas | 36% (n = 14) |
| Kavuru et al.39 | GM-CSF sc | 250μg/día hasta 5-9μg/kg/día | 12 semanas | 75% (n = 4) |
| Bonfield et al.30 | GM-CSF sc | 250μg/día hasta 18μg/kg/día | 12-48 semanas | 55% (n = 11) |
| Venkateshiah et al.31 | GM-CSF sc | 250μg/día hasta 5-18μg/kg/día | 12-52 semanas | 48% (n = 21) |
| Tazawa et al.32 | GM-CSF inh | 250μg/día semanas alternas | 24 semanas | 100% (n = 3) |
| Wylam et al.33 | GM-CSF inh | 250-500μg/día | 12 semanas | 83% (n = 12) |
| Borie et al.34 | Rituximab iv | 1g día 0 y 15 | 15 días | 100% (n = 1) |
| Amital et al.35 | Rituximab iv | 375mg/m2 semanal | 4 semanas | 100% (n = 1) |
| Kavuru et al.36 | Rituximab iv | 1g días 0 y 15 | 15 días | 78% (n = 9) |
inh: vía inhalatoria; iv: vía intravenosa; sc: vía subcutánea.
Kavuru et al.29 trataron a un grupo de 4 pacientes durante 12semanas. En las primeras 4semanas la dosis fue de 250μg/día, incrementándose 5μg/kg/día las siguientes 4semanas y finalmente 9μg/kg/día las últimas 4semanas. En 3 de los 4 casos se produjo mejoría de la función pulmonar, la oxigenación y en la capacidad de esfuerzo.
En un ensayo prospectivo, Bonfield et al.30 incluyeron 14 casos de PAP, de los que 11 completaron el estudio. Todos los casos habían sido tratados previamente con lavado pulmonar total. La dosis inicial fue de 250μg/día, incrementando la dosis cada 2semanas hasta un máximo de 18μg/kg/día. El tiempo total de tratamiento fue de 12-48semanas. En 6 casos se produjo una mejoría en la oxigenación, pasando de 69,1±3,4mmHg a 84,0±2,0mmHg. En el grupo que respondió al tratamiento se produjo un descenso en los niveles de anti GM-CSF tanto en suero como en BAL.
En un ensayo clínico abierto prospectivo con 25 pacientes31, 21 lo completaron. Hubo una mejoría significativa en 12 casos (57%), y en 6 de estos (67%) no fueron necesarias la realización de lavado pulmonar total ni la administración de oxígeno domiciliario.
A pesar de las limitaciones metodológicas, las diferentes dosis administradas y la duración variable de los tratamientos, la administración de GM-CSF por vía subcutánea parece eficaz hasta en 2/3 de los casos, con pocos efectos secundarios que incluyen edema y eritema en el sitio de punción y febrícula. No suelen apreciarse alteraciones en las células sanguíneas. Supone una alternativa al tratamiento de la PAP.
GM-CSF por vía inhaladaTazaba et al. publicaron una serie de 50 pacientes con PAP primaria32 a la que se administró GM-CSF por vía inhalatoria a una dosis de 125μg cada 12h durante una semana, con otra de descanso, haciendo en total 6 ciclos de 2semanas (12semanas de tratamiento total) y con un seguimiento posterior de 52semanas tras la finalización del tratamiento. De los 35 pacientes que completaron el ensayo, en 24 hubo una clara mejoría en la diferencia alveoloarterial de oxígeno (12,3mm Hg, IC95% 8,4-16,2mm Hg, p<0,001), con una mejoría global del 62% (en 24/39 pacientes en el análisis final por intención de tratar). Durante el seguimiento, en 29 de los 35 pacientes se constató estabilidad clínica, sin precisar ningún otro tipo de tratamiento adicional.
Wylam et al.33 llevaron a cabo un estudio retrospectivo en 12 pacientes con PAP primaria con la administración de GM-CSF en aerosol con dosis crecientes hasta un máximo de 500μg/día durante un período de 12semanas. Los resultados fueron positivos, alcanzando mejoría en 11 pacientes en síntomas, y en 10 de ellos hubo mejoría significativa en el intercambio gaseoso. Los resultados a largo plazo se mantuvieron, alcanzando en 8 casos una remisión parcial, y en 3 se consiguió una remisión completa radiológica.
El tratamiento con GM-CSF por vía inhalatoria podemos considerar que es eficaz en 4/5 de los casos, con pocos efectos secundarios. Las dosis empleadas y la duración de los tratamientos son variables.
RituximabEste anticuerpo monoclonal está dirigido contra el receptor antigénico CD20 situado en la superficie de los linfocitosB. Esta diana es una proteína de membrana de 35kD que tiene un papel fundamental en el ciclo celular y la diferenciación de los linfocitosB34. La administración de rituximab produce un descenso rápido de los linfocitosB en sangre periférica. Se han propuesto varios mecanismos de acción: citotoxicidad mediada por el complemento, citotoxicidad mediada por células dependiente de anticuerpos o incremento de la apoptosis mediada por anticuerpos. Rituximab se emplea en el tratamiento de enfermedades linfoproliferativas de linfocitosB y en varias enfermedades autoinmunes, como lupus eritematoso sistémico, artritis reumatoide o vasculitis35. Dado que la PAP primaria es considerada una enfermedad autoinmune en la que participan anticuerpos frente al GM-CSF, la reducción de los linfocitosB y los niveles de anti GM-CSF que produciría rituximab podría ser una opción eficaz para el tratamiento36.
Borie et al.37 describieron el efecto de rituximab en un paciente con PAP que rechazó la realización de lavado pulmonar. Se administraron 2dosis de 1g con 15días de diferencia, y se comprobó un descenso del número de linfocitosB y los niveles de anti GM-CSF en suero. A los 6meses del tratamiento el gradiente alveoloarterial había mejorado, pero no se produjo ningún cambio en la DLCO ni en los hallazgos del TCAR. Amital et al.38 describen el efecto de rituximab en una paciente con PAP que no había conseguido mejorar tras la realización de un lavado pulmonar total y la posterior administración de GM-CSF por vía subcutánea. La dosis fue de 375mg/m2 semanal durante 4semanas, consiguiendo mejorar el intercambio gaseoso, la DLCO y los hallazgos radiológicos.
El estudio más amplio publicado es un ensayo clínico abierto con rituximab en 10 pacientes con PAP39. Se administraron 2dosis de 1g en el día 0 y el día 15. La diferencia alveoloarterial de O2 y la PaO2 mejoraron en 7 de 9 pacientes que completaron el ensayo y se mantuvo a los 3 y 6meses de haber completado el tratamiento. Hubo mejoría también en los hallazgos del TCAR y en la función pulmonar.
El tratamiento con rituximab no está exento de efectos secundarios, algunos de ellos graves, y a pesar de ser un tratamiento prometedor continúa siendo una opción terapéutica para la PAP que está en el campo de la investigación.
Actualmente hay varios ensayos clínicos activos en los que se evalúan tanto rituximab como GM-CSF en la PAP; información disponible en http://apps.who.int/trialsearch/.
PlasmaféresisEl tratamiento con plasmaféresis para disminuir los niveles de anti GM-CSF en suero se ha intentado en varios casos aislados en los que otros tratamientos, como lavado pulmonar, anti GM-CSF o rituximab, no habían sido eficaces. Los resultados han sido dispares, y su eficacia como tratamiento de la PAP actualmente no está establecida40,41.
Trasplante pulmonarSe han realizado trasplantes pulmonares en aquellos casos en los que los tratamientos no han sido eficaces y la enfermedad ha progresado. Se han comunicado casos aislados de trasplante pulmonar en adultos, y en una serie de 270 trasplantes realizados en 190 niños entre 1990 y 2002, en 12 casos (6,3%) se hicieron por PAP42. Es una opción para aquellos casos graves y refractarios al tratamiento. Se ha descrito la recurrencia de la PAP a los 3años en el pulmón trasplantado43, pero se desconoce la frecuencia de la posible recurrencia. Posiblemente con el desarrollo de las nuevas terapias para la PAP, el trasplante puede ser una opción para casos muy excepcionales.
CorticoidesNo están indicados en el tratamiento de la PAP, y de hecho pueden empeorar el pronóstico, ya que pueden interferir con el metabolismo del surfactante44 y alterar la respuesta inmune4. En los primeros trabajos publicados sobre PAP se describieron casos con desenlace fatal asociado a desarrollo de infecciones por Nocardia, criptococo o mucormicosis en algunos casos que habían sido tratados con corticoides45.
Trasplante alogénico de precursores hematopoyéticosEn un modelo murino se ha conseguido revertir la PAP tras el trasplante de progenitores hematopoyéticos, y se han publicado casos de PAP secundaria o hereditaria con buena evolución tras este tratamiento5. Sin embargo, es un tratamiento aun en investigación y del que existe poca evidencia. Una nueva línea de investigación, que evitaría el tratamiento ablativo previo, podría ser el trasplante pulmonar de macrófagos alveolares46, que ha mostrado resultados prometedores en modelos murinos y que podría ser una opción de tratamiento en un futuro47.
Por último, estos pacientes pueden sufrir infecciones bacterianas que requieren tratamiento antibiótico adecuado. Existe una mayor susceptibilidad por el defecto en la actividad macrofágica a las infecciones por gérmenes como Nocardia o P.jirovecii. Por este motivo, algunos autores recomiendan el tratamiento profiláctico con trimetroprim/sulfametoxazol hasta conseguir la remisión completa de la enfermedad48.
ConclusiónEl tratamiento de elección para la PAP sigue siendo el lavado pulmonar total, pero no se requiere su realización en todos los pacientes. Con el mayor conocimiento de los mecanismos patogénicos de esta enfermedad se han incrementado las opciones terapéuticas (fig. 2). En la PAP primaria o autoinmune se ha utilizado con éxito el GM-CSF, tanto inhalado como subcutáneo, pero la dosis óptima a emplear, la duración del tratamiento y la vía de administración del GM-CSF no se han establecido definitivamente. Existen otros tratamientos prometedores pero que aún están en fase de investigación. Debido al escaso número de casos de esta enfermedad, sin olvidar la posibilidad de su remisión espontánea, es difícil establecer tratamientos con nivel de evidencia alto. Para los casos más graves, posiblemente en el futuro emplearemos terapias combinadas.
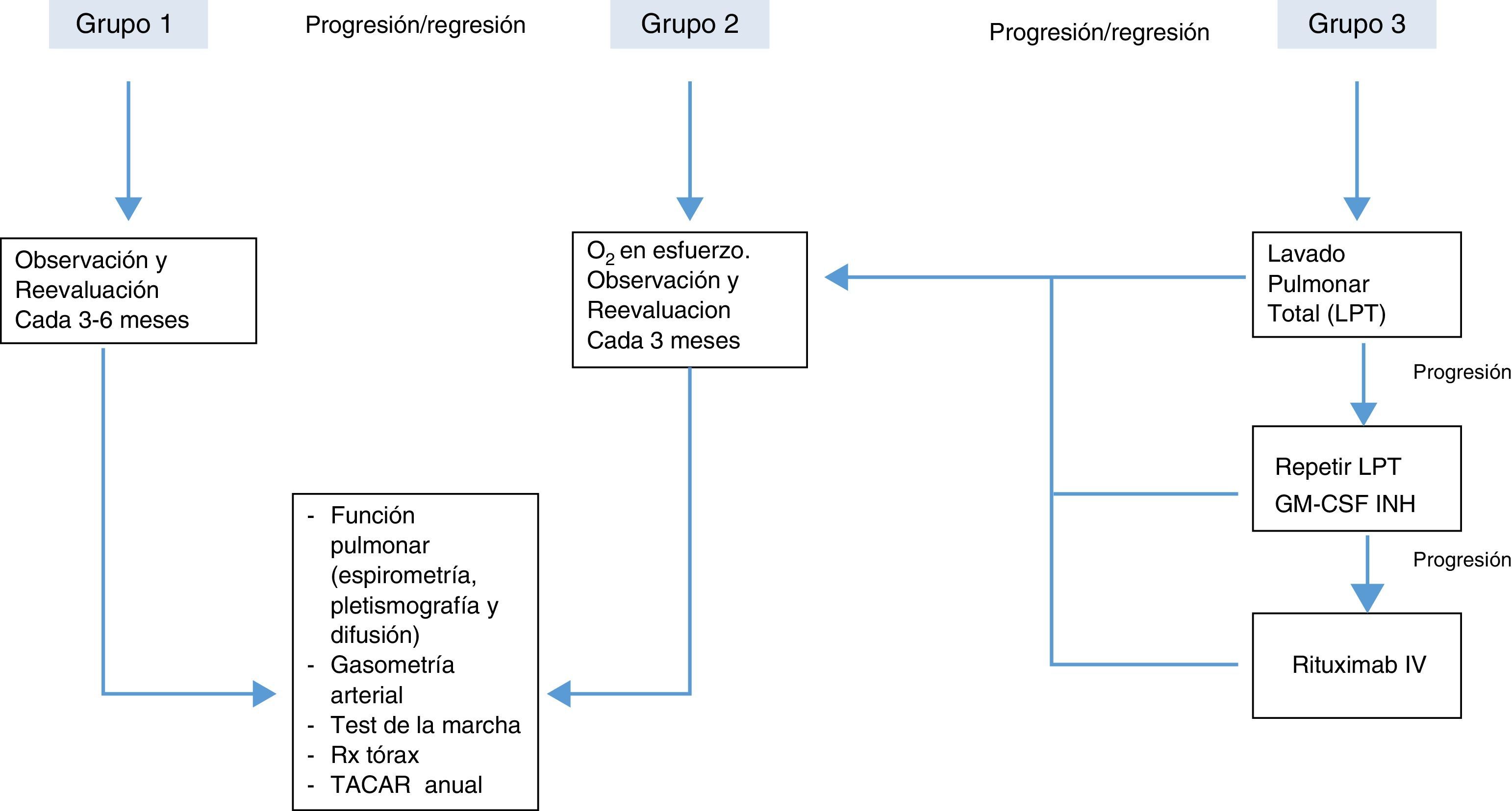
Posible algoritmo de tratamiento de la PAP según gravedad, basado en las opiniones de los diferentes autores20.
Los autores declaran no tener ningún conflicto de intereses.







