



Thymic carcinoids are a rare entity that may be associated with endocrine diseases like Cushing's syndrome or multiple endocrine neoplasia syndrome type I (MEN1). These tumors represent 4% of anterior mediastinal tumors and are characterized by their very aggressive behavior.
We present the case of a patient with a previous MEN 1 diagnosis in whom, during the follow up of his disease, a thoracic image compatible with thymic carcinoid was detected. After an extended thymectomy that included peri-thymic fat resection, the clinical diagnosis was confirmed. A follow-up examination 14 months later revealed a suspicious lesion that suggested local recurrence, therefore the patient was reoperated on. The pathology report of this surgery indicated post-radiation fibrosis.
Likewise, we present a review of the current diagnostic and therapeutic management of patients with MEN1 syndrome who are diagnosed with thymic carcinoid.
Los carcinoides tímicos son una entidad poco frecuente que puede asociarse a enfermedades endocrinológicas como el síndrome de Cushing o el síndrome de neoplasia endocrina múltiple tipo I (MEN1). Suponen el 4% de los tumores del mediastino anterior y se caracterizan por tener un comportamiento muy agresivo.
Presentamos el caso de un paciente diagnosticado de síndrome MEN1 a quien durante el seguimiento de su enfermedad se detectó una imagen torácica compatible con carcinoide tímico. Tras intervenirle quirúrgicamente mediante timectomía ampliada a grasa peritímica, se confirmó el diagnóstico clínico. A los 14 meses de seguimiento se halló en las pruebas de imagen una lesión sospechosa de recidiva local, motivo por el que fue reintervenido. El informe anatomopatológico de dicha intervención fue de fibrosis rádica.
Así mismo, presentamos una revisión del manejo diagnóstico y terapéutico actual en pacientes con síndrome MEN1 diagnosticados de carcinoide tímico.
Thymic carcinoid tumors were described for the first time by Rosai and Higa in 1972 as an entity independent from thymic carcinomas.1 They are neuroendocrine tumors derived from the APUD cells of the thymus and represent 4% of the tumors of the anterior mediastinum, with a predilection for men aged 30–60.2 Clinically, these patients can either be asymptomatic or show symptoms of pain or compression, and may be associated with syndromes like Cushing's in up to 40% of cases or multiple endocrine neoplasia syndrome type 1 (MEN1) in up to 25% of cases, among other entities.3–5 It is characterized by a very aggressive behavior, frequently presenting local invasion upon diagnosis. Lymph node invasion and distance metastasis can be observed in 50% and 20% of cases, respectively.6
Case ReportWe present the case of a 43-year-old male patient with a personal history of MEN 1 syndrome and giant pituitary adenoma (which had been treated on 2 occasions surgically and with adjuvant radiotherapy), primary hyperparathyroidism requiring total parathyroidectomy with autotransplantation in the left forearm with anatomic pathology results for hyperplasia and an area of adenoma, as well as a gastrinoma being treated with somatostatin analogues. Secondary to the latter, the patient presented a perforated bile duct ulcer that required Billroth II gastrectomy. In March 2006, follow-up octreotide scintigraphy of the gastrinoma showed pathologic uptake in the anterior mediastinum, and the patient was therefore sent for consultation with the thoracic surgery department for assessment given the suspicion for a second primary tumor.
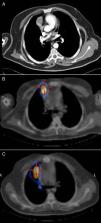
Upon physical examination, the patient was asymptomatic. Workup values showed: prolactin, 139ng/ml; total testosterone total 1.16ng/ml; free testosterone 3.41pg/ml; PTH 2.112pg/ml; gastrin, >1050pg/ml; calcium, 11.2mg/dl; phosphorus 2.82mg/dl. Computed tomography (CT) revealed a 3-cm solid mass with intravenous contrast uptake in the anterior mediastinum in contact with the pleura and right pericardium near the homolateral auricle, with no calcifications or cavitations (Fig. 1A). Octreotide scan detected 2 nodular lesions with elevated high somatostatin receptor density: one in the right hemithorax and the second at the tail of the pancreas. SPECT/CT confirmed the location of both lesions (Fig. 1B).

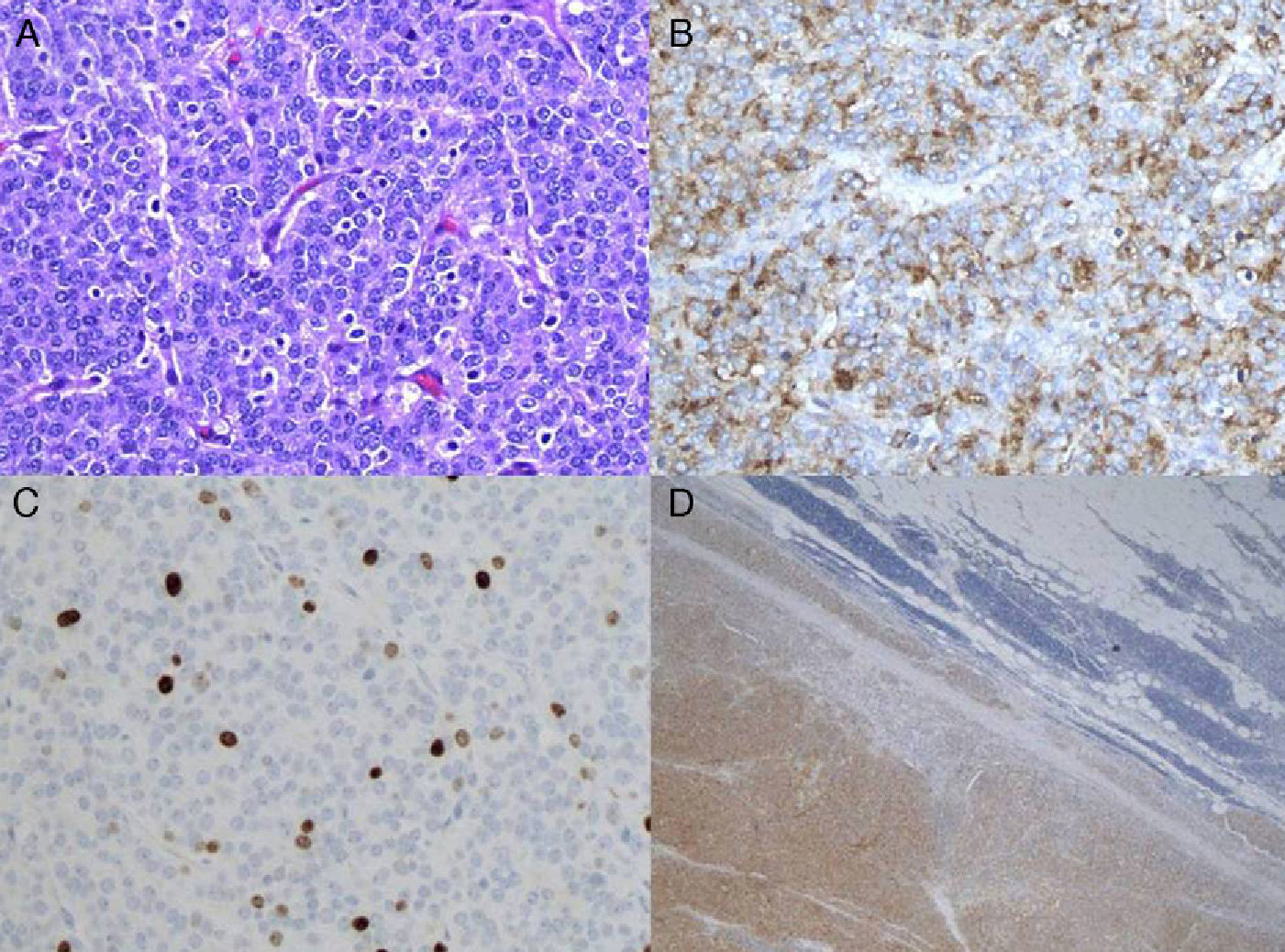
In August 2006, the patient underwent extended thymectomy by median sternotomy. A thymic tumor was observed, dependent on the right lobe, measuring 5cm in diameter and with no infiltration of mediastinal structures. Total thymectomy was carried out and extended to the right mediastinal pleura. The anatomic pathology study diagnosed thymic carcinoid tumor (Fig. 2A–D), classified as Masaoka stage II and pT1N0M0 stage I. The post-operative period transpired without incident and the patient was discharged four days after surgery. In October 2006, adjuvant mediastinal radiotherapy was initiated at a dosage of 56Gy with no complications.
During outpatient follow-up, SPECT/CT in October 2007 showed increased uptake in the right paramediastinal region (Fig. 1C). CT confirmed the presence of an anterior mediastinal consolidation with pseudo-nodular morphology in contact with the pleura and the right pericardium and proximal to the ipsilateral auricle. In February 2008, the patient once again underwent surgery, specifically right thoracotomy, with no observation or palpation of any nodules; therefore, part of the pericardium was resected as well as the area of fibrosis that had been theoretically located by increased uptake during scintigraphy. The anatomic pathology study reported no evidence of neoplasm and the diagnosis was radiation fibrosis. Currently, the patient is disease-free and continues in oncologic follow-up including annual CT.
In January 2010, the brother of the patient was diagnosed with the same syndrome after hyperparathyroidism. When he underwent the appropriate parathyroidectomy, prophylactic thymectomy was also performed.
DiscussionWhen treating patients diagnosed with MEN 1 syndrome, it is important to rule out the presence of thymic carcinoid tumor due to the fact that these two entities may be associated in up to 25% of cases. For the screening and follow-up of these patients, annual chest radiography and chest CT every 3 years have been proposed.7,8
The diagnosis of thymic carcinoid tumors should be based on chest CT, extended to include the upper part of the abdomen in order to rule out the existence of a possible secondary affectation. These tumors typically present as irregular masses that may contain calcifications and are enhanced with intravenous contrast.9,10 During progression, they may show signs of local invasion and the pleura, pericardium and lung parenchyma are the most frequently compromised structures. Octreotide scan is a more specific test that can also help to detect distant metastasis, and it is a follow-up method used when positive in the pre-operative work-up.11
Other tests with which there has been less experience are magnetic resonance imaging (MRI) (which is especially useful for the assessment of locoregional invasion of mediastinal structures), single-photon emission computed tomography (SPECT) and positron emission tomography (PET).11 Although the experience with PET in thymic carcinoid tumors is limited, its usefulness in the case of invasive thymomas and thymic carcinomas seems to have already been demonstrated, which would theoretically support its use in the evaluation of aggressive tumors like thymic carcinoids.12 New tracers are being studied in order to more precisely diagnose carcinoid tumors, especially thymic carcinoids.13
Whenever possible, the treatment of choice is surgery and the technique of choice is en bloc resection of the thymus with any adjacent compromised structures.14 It may be associated with neoadjuvant or adjuvant chemotherapy and/or radiotherapy, although it is yet to be established which treatment is best as existing studies are based on series with few patients. As for radiotherapy, studies have been published that reflect its usefulness in the prevention of relapse.6,14–16 Likewise, it is probable that chemotherapy should be systematically considered due to the high rate of distant metastasis (20%).3 Another therapeutic option is hormonal therapy with octreotide, given its good antiproliferative activity in neuroendocrine tumors, even in cases of patients with distance metastasis.17 In using this option, is it recommended to determine somatostatin receptors by octreotide scan.18 A case has been published of successful multidisciplinary treatment using induction chemoradiotherapy, surgery and adjuvant hormone therapy.3
In our case, as there was no pre-operative histologic diagnosis of the lesion, the patient was directly treated with en bloc thymectomy extended to the mediastinal pleura. As adjuvant treatment, we opted for radiotherapy and somatostatin analogues, and the patient remained disease-free after 52 months of follow-up.
As for the factors related with greater relapse, incomplete resection and advanced stage disease are most frequently reported.19 Survival in the Gal et al. series varied according to resectability: for patients with complete resection, 5-year survival was 77% and 10-year was 30%; for partial resections, 5- and 10-year survival rates were 65% and 19%, respectively; and, for no resection, they were 28% and 0%, respectively.19 Moran confirmed said results with 5- and 10-year survival rates of 28% and 10%, respectively.2 Another factor involved in prognosis is the degree of tumor differentiation: for low-grade tumors, mean survival ranged from 9 to 11 years; for intermediate-grade tumors, this rate ranged from 5 to 7 years; for high-grade tumors, mean survival varied from 18 to 3 years.2,19
Regarding preventive measures, the option of carrying out a prophylactic thymectomy simultaneously with total or subtotal parathyroidectomy should be contemplated.20 This reduced the risk for relapse of hyperparathyroidism as a consequence of ectopic supernumerary glands, as well as the risk for developing associated thymic carcinoid tumors.
Conflict of InterestsThe authors declare having no conflict of interests.
Please cite this article as: Recuero Díaz JL, et al. Carcinoide tímico asociado a síndrome de neoplasia endocrina múltiple tipo I. Arch Bronconeumol. 2013;49:122–5.







