






Cystic fibrosis neonatal screening (CFNS), based on double determination of immunoreactive trypsinogen ([IRT] [IRT1/IRT2]), has been available in Andalusia since May 2011. If screening is positive, a sweat test is performed, and if that is positive or inconclusive, genetic testing is requested.
ObjectiveTo analyze CFNS, based on results from the first 4.5 years of the program.
Materials and methodsProspective descriptive study of neonates undergoing CFNS. IRT levels, sweat chloride, and mutations were recorded. Statistical analysis was performed using SPSS 12.0.
ResultsBetween May 2011 and December 2016, 474,953 neonates underwent CFNS. Of these, 1087 (0.23%) had elevated IRT2. Since CFNS was introduced, 73 cases of cystic fibrosis were diagnosed; 60 were diagnosed by positive CFNS, and 13 were diagnosed by other means. In one case, the patient developed a typical clinical picture of cystic fibrosis, but had not undergone CFNS at the decision of the parents; the remaining 12 had a negative CFNS (false negatives). Of these, one patient was diagnosed before symptoms developed, as his twin brother had a positive CFNS result; another had chloride at the upper limit of normal, and was subsequently diagnosed with genetic testing before symptoms appeared; and 10 patients developed clinical signs and symptoms. Excluding patients with meconium ileus, sensitivity and specificity of the CFNS program were 85.71% and 99.78%, respectively. The incidence of the disease in Andalusia is 1/6506 live births.
ConclusionThese results are a basis for reflection on possible areas for improvement of the CFNS algorithm, and thought may be given to the introduction of genetic studies to increase sensitivity and reduce false positives.
Andalucía dispone de screening neonatal de fibrosis quística (SNFQ) desde mayo 2011, basado en doble determinación de tripsinógeno inmunorreactivo ([TIR] [TIR1/TIR2]). Si el screening es positivo realizamos un test del sudor y si es positivo o dudoso solicitamos genética.
ObjetivoAnalizar el SNFQ, basado en los resultados de los primeros 4,5 años.
Material y métodoEstudio descriptivo prospectivo de los neonatos sometidos a SNFQ. Se recogen los niveles de TIR, cloruro en sudor, mutaciones. Mediante SPSS12.0 se realizó análisis estadístico.
ResultadosDesde mayo 2011 a diciembre 2016, 474.953 neonatos fueron sometidos a SNFQ. Mil ochenta y siete (0,23%) presentaron TIR2 elevado. Desde la implantación del SNFQ se diagnosticaron 73 casos con fibrosis quística; 60 de ellos fueron diagnosticados mediante un SNFQ positivo, mientras que 13 no. Concretamente un paciente comenzó con clínica clásica de fibrosis quística y se comprobó que no se había realizado el SNFQ por decisión paterna; los 12 restantes tuvieron un SNFQ negativo (falsos negativos). De estos, un paciente fue diagnosticado presintomáticamente al tener su hermano gemelo con SNFQ positivo; otro con cloruro en el límite alto de la normalidad se diagnosticó presintomáticamente mediante genética; 10 pacientes comenzaron clínicamente. Excluyendo los pacientes con íleo meconial, la sensibilidad y especificidad del programa de SNFQ asciende al 85,71 y 99,78% respectivamente. La incidencia de la enfermedad en Andalucía es de 1/6.506 recién nacidos vivos.
ConclusiónLos presentes resultados nos permiten reflexionar sobre posibles áreas de mejoras adicionales del algoritmo del SNFQ, que debe pasar por la introducción de estudios genéticos para así aumentar la sensibilidad y disminuir los falsos positivos.
Cystic fibrosis (CF, OMIM #219700) is a chronic, multisystem disease associated in most cases with respiratory infections, pancreatic failure, and increased chloride levels in sweat, caused by mutations in the cystic fibrosis transmembrane conductance regulator gene (CFTR).1–3 Over 2000 genetic variants have been identified to date.1,2,4 We are currently witnessing a significant increase in survival, thanks to improvements in nutrition, better access to systemic and inhaled antibiotic therapy, use of pancreatic enzymes, improved lung transplantation procedures, treatment with CFTR modulators, and the introduction of neonatal screening programs.1,2,5–7
There is currently no universally accepted protocol for CF neonatal screening (CFNS), but all programs use immunoreactive trypsinogen (IRT) quantification as an initial marker3,8,9 IRT levels are elevated in most CF patients, but they can also rise for other reasons, including prematurity and low birth weight, producing false positives and generating high levels of anxiety among families.1,2,8 To increase specificity, a second marker, such as pancreatitis-associated protein (PAP), a second IRT quantification (IRT2), or determination of CFTR mutations, is generally determined.8,9,13
No data are available yet on the most suitable CF screening protocol. If we are to develop a consensus strategy in the future, the first step must be to examine the results of current screening programs in order to determine the yield of the various algorithms.
CFNS was introduced in Andalusia in May 2011, as part of the expanded neonatal screening program for 30 diseases. The aims of this study were to evaluate the results of the Andalusian CFNS program on the basis of results from the first 4.5 years, to describe its strengths, to identify areas for possible improvement, and to compare or extrapolate the results to other communities or countries using the same CFNS protocol.
Materials and MethodsThis was an observational, cross-sectional study that included all infants born in Andalusia between May 1, 2011 and December 31, 2016. Newborn screening procedures are centralized in 2 reference units: the Hospital Universitario Virgen del Rocío in Seville and the Hospital Regional Universitario Carlos Haya in Malaga. Blood samples were collected on filter paper with the heel stick method, using Perkin Elmer 226 collection devices (Waltham, MA, USA), within 48–72h after birth. These dry blood samples are sent by post to the 2 neonatal screening reference units in Andalusia, according to geographical distribution. These samples undergo immunoanalysis using the AutoDELFIA Neonatal IRT kit for IRT determination.
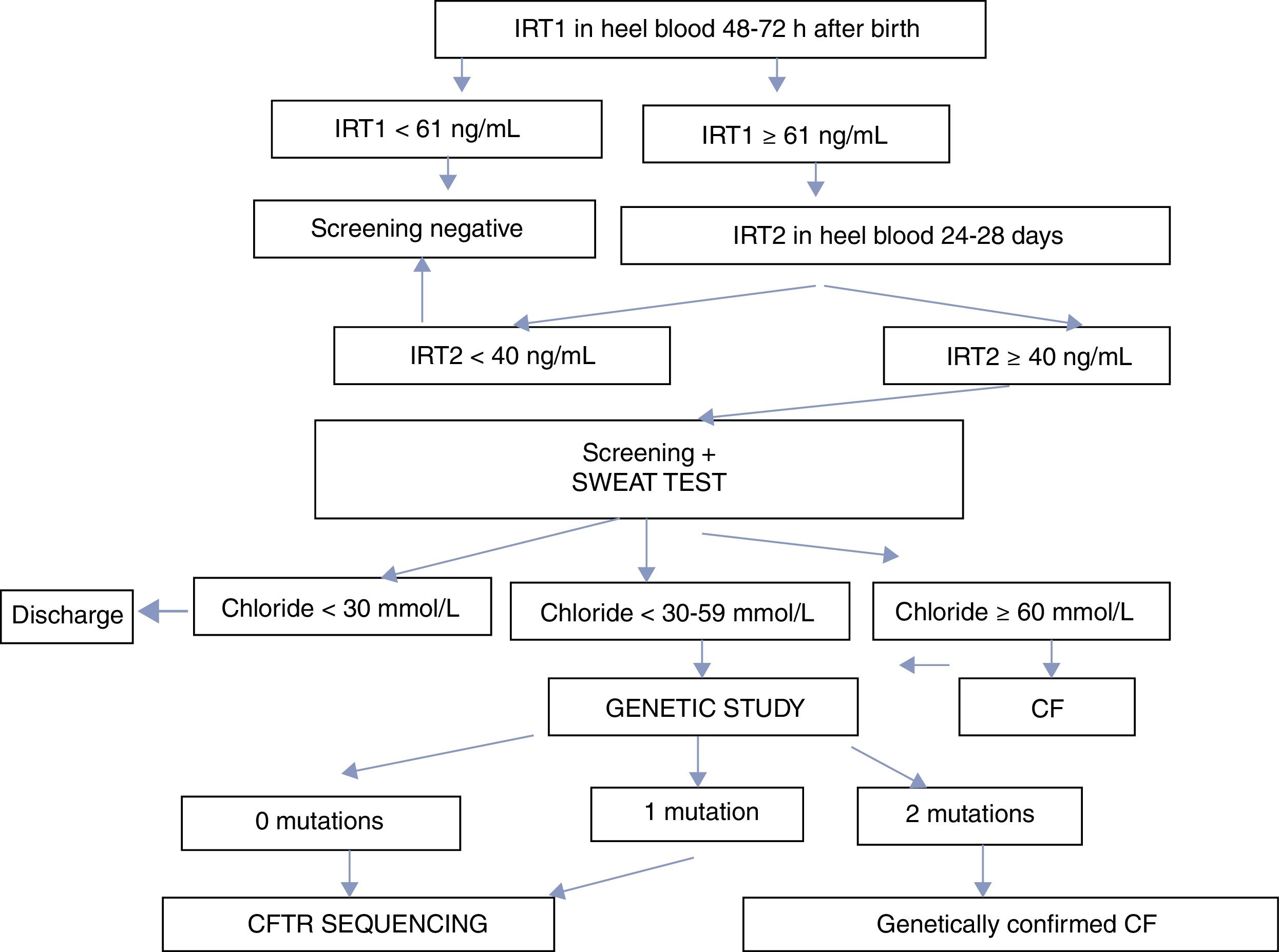
The screening program is based on the double determination of IRT. The first IRT (IRT1) is quantified within 48–72h after birth. If values are ≥61ng/mL, a second determination is requested between days 25 and 28 after birth. If IRT2 is ≥40ng/mL, the infant is referred to a reference unit for a sweat test. If the sweat test is equivocal or positive (chloride ≥30mmol/L), a genetic study is performed. The CFNS program in Andalusia is summarized in Fig. 1.
Sweat tests were performed on at least 2 occasions with iontophoresis with pilocarpine, using a Macroduct® collection device (Wescor, Logan, UT, USA), and chloride was determined in a MK II Chloride Analyzer 926S (Sherwood Scientific, Ltd).
Confirmatory genetic tests were conducted to validate the results of IRT and sweat testing. First, the patient's DNA was automatically extracted from a sample of peripheral blood (MagNA Pure Compact, Roche, Germany). In the first step, the Elucigene CF-EU2 kit (Elucigene Diagnostics, UK), that can detect 50 mutations simultaneously using Amplification Refractory Mutation System (ARMS) technology, was employed. Generated fragments were analyzed by capillary electrophoresis in the ABI3730 sequencer using GeneMapper v 4.0 software (ThermoFisher Scientific, Waltham, MA, USA). In cases with equivocal or positive sweat tests in which the 2 mutations were not identified among the panel of 50 mutations, full sequencing of the CFTR gene was performed by massive sequencing, using the Multiplicom MASTRTM Dx CFTR kit and the MiSeq® sequencer (lllumina). Massive sequencing data were analyzed using SeqNext module of the SEQUENCE Pilot software (JSI Medical Systems).
The study met the ethical requirements of the Declaration of Helsinki on studies involving human subjects and the Spanish regulations on data protection and confidentiality (Act 15/1999 of 13 December on personal data protection). Clinical records were anonymized in the database with a numerical code assigned by an algorithm. No personal data that could be used directly or indirectly to identify an individual were entered. The link between the database code and the clinical record number was stored locally under the responsibility of the on-site investigator. Informed consent was obtained from the parents in all cases in which genetic studies were performed.
Statistical AnalysisThe statistical analysis was carried out with SPSS12.0 software. After confirmation of the screening and genetic diagnostic results, the definitive diagnosis was assessed with calculation of sensitivity and specificity profiles and predictive values with confidence intervals. The level of statistical significance was set at 0.05.
ResultsBetween May 1, 2011 and December 31, 2016, a total of 474,953 newborns underwent CFNS with IRT1 determined using the AutoDELFIA system. An overview of the diagnostic algorithm and the results are presented in Fig. 1 and Table 1, respectively. The trend analyses of the percentiles were reviewed on a monthly basis. The limit of IRT1 was initially established at 65ng/mL and IRT2 at 35mmol/L, until November 2012, when they were adjusted to 60 and 40ng/mL, respectively, representing the 99.5th percentile of accumulated IRT at that point.
Summary of Characteristics of all Patients Diagnosed with CF Since the Implementation of the CFNS.
| Sex | Days at Diagnosis | IRT1 (ng/ml) | IRT2 (ng/ml) | 1st Chloride in Sweat (mmol/l) | 2nd Chloride in Sweat (mmol/l) | Mutation 1 | Mutation 2 | Pancreatic Sufficiency. (0=PI; 1=PS) | Diagnosis | Diagnostic Symptoms of FN Observations | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 15 | 112 | – | 103 | IM | F508del | F508del | 0 | TP | Meconium ileus |
| 2 | M | 600 | – | – | 55 | 60 | c.2657+5G>A | Unknown | 1 | FN | Heat prostration |
| 3 | M | 87 | 120 | 59 | 42 | IS | F508del | D1152H | 1 | TP | |
| 4 | M | 52 | 115 | 69 | 79 | IS | R334W | Q1281X | 1 | TP | |
| 5 | M | 34 | 218 | 369 | 103 | IS | F508del | IVS18-1G>A | 0 | TP | |
| 6 | M | 51 | 140 | 73 | 81 | IS | F508del | G461R | 1 | TP | |
| 7 | M | 42 | 151 | 114 | 60 | 56 | G542X | G461R | 1 | TP | |
| 8 | M | 35 | 86 | 123 | 81 | IS | F508del | G542X | 0 | TP | |
| 9 | M | 34 | 182 | 152 | 100 | IS | F508del | I507del | 0 | TP | |
| 10 | M | 34 | 139 | 128 | 65 | IS | F508del | F508 | 0 | TP | |
| 11 | M | 40 | 112 | 149 | 93 | IS | G85E | Q493X | 1 | TP | |
| 12 | M | 600 | IS | 7 | 112 | IS | p.Leu997Phe | c.2909-92G>A | 0 | FN | Typical clinical picture |
| 13 | M | 43 | 194 | 92 | 73 | IS | F508del | F508del | 0 | TP | |
| 14 | M | 31 | 153 | 144 | 85 | IS | F508del | G542X | 0 | TP | |
| 15 | M | 43 | 92 | 175 | 115 | IS | F508del | p.Tyr515X | 0 | TP | |
| 16 | M | 15 | 331 | – | 94 | IS | F508del | F508del | 0 | TP | Meconium ileus |
| 17 | M | 146 | 89 | 34 | 87 | IS | F508del | F508del | 0 | FN | Typical clinical picture |
| 18 | M | 60 | 97 | 161 | 110 | IS | F508del | F508del | 0 | TP | |
| 19 | M | 34 | 61 | 125 | 90 | IS | F508del | F508del | 0 | TP | |
| 20 | M | 240 | 48 | – | 73 | IS | N1303K | R117C | 1 | FN | Dehydration |
| 21 | M | 25 | 62 | 107 | 82 | IS | F508del | F508del | 0 | TP | Meconium ileus |
| 22 | M | 36 | 187 | 60 | 101 | IS | F508del | W1282X | 0 | TP | |
| 23 | M | 41 | 126 | 386 | – | IS | F508del | p.Glyn493ValfsX10 | 0 | TP | |
| 24 | M | 48 | 66 | 77 | 77 | IS | p.Arg1162X | Unknown | 1 | TP | |
| 25 | M | 32 | 122 | 160 | 90 | IS | F508del | F508del | 0 | TP | |
| 26 | M | 30 | 232 | 121 | 84 | IS | I507del | Unknown | 0 | TP | |
| 27 | M | 40 | 96 | 88 | IS | IS | F508del | W1282X | 0 | TP | |
| 28 | M | 65 | 200 | 310 | 79 | IS | c.579+5G>A | c.2988+1KbDel8.6Kb | 0 | TP | |
| 29 | M | 19 | 85 | 145 | 90 | IS | c.579+1G>T | Q890X | 0 | TP | Meconium ileus |
| 30 | M | 58 | 263 | 300 | IS | IS | F508del | c.1585-1G>A | 0 | TP | |
| 31 | M | 58 | 161 | 254 | IS | IS | F508del | c.1585-1G>A | 0 | TP | |
| 32 | M | 61 | 140 | 178 | IS | IS | F508del | S549R | 0 | TP | |
| 33 | M | 51 | 147 | 211 | 116 | IS | F508del | c.1585-85>A | 0 | TP | |
| 34 | M | 59 | 103 | – | 120 | 93.7 | F508del | G542X | 0 | TP | |
| 35 | M | 58 | 63.6 | 44.6 | 119 | 106.1 | N1303K | R334W | 1 | TP | |
| 36 | M | 58 | 61.4 | 41.3 | 113 | 101.8 | N1303K | R334W | 1 | TP | |
| 37 | M | 42 | 75.6 | 63 | 99 | 76 | F508del | c.2657+5G>A | 1 | TP | |
| 38 | M | 42 | 39.3 | – | 98 | 72.5 | F508del | c.2657+5G>A | 1 | FN | Twin brother with screening+ |
| 39 | M | 34 | 133 | 113 | 122 | 92.9 | R334w | S549R | 1 | TP | |
| 40 | M | 35 | 85.6 | 115 | 102 | 109 | F508del | G542X | 0 | TP | |
| 41 | M | 32 | 101 | 79 | – | – | F508del | R334W | 1 | TP | |
| 42 | M | 35 | 148 | 175 | 112 | IS | F508del | G542X | 0 | TP | |
| 43 | M | 33 | 93.7 | 233 | 100 | 77.1 | F508del | F508del | 0 | TP | |
| 44 | M | 36 | 58.4 | 52.4 | 78 | 81 | F508del | R334W | 1 | TP | |
| 45 | M | 34 | 212 | 165 | 101 | 98 | F508del | S1196X | 0 | TP | |
| 46 | M | 45 | 230 | 496 | 109 | 107 | F508del | F508del | 0 | TP | |
| 47 | M | 180 | 33 | – | 59 | 57 | F508del | c.3717+10kbC>T | 1 | FN | Heat prostration |
| 48 | M | 36 | 90.7 | 45.70 | 89 | 96 | F508del | R334W | 1 | TP | |
| 49 | M | 34 | 176 | 70.30 | 101 | 110 | F508del | F508del | 0 | TP | |
| 50 | M | 14 | 41 | – | 74 | – | p.Ile507del | F508del | 0 | FN | Meconium ileus |
| 51 | M | 36 | 105 | 70.90 | 29 | 32 | F508del | c.2657+5G>A | 1 | TP | |
| 52 | M | 36 | 94.9 | 62.10 | 64 | 69 | F508del | c.2657+5G>A | 1 | TP | |
| 53 | M | 32 | 132 | 210 | 105 | 79.4 | F508del | F508del | 0 | TP | |
| 54 | M | 36 | 120 | 448 | 112 | 94.7 | F508del | F508del | 0 | TP | |
| 55 | M | 32 | 132 | 422 | 87 | IS | F508del | F508del | 0 | TP | |
| 56 | M | 35 | 239 | 287 | 118 | 103 | F508del | F508del | 0 | TP | |
| 57 | M | 35 | 151 | 179 | 104 | 98 | F508del | G542X | 0 | TP | |
| 58 | M | 33 | 169 | 169 | 100 | 91.1 | F508del | F508del | 0 | TP | |
| 59 | M | 36 | 122 | 82.90 | 105 | 101 | c.1680-1G>A | G542X | 0 | TP | |
| 60 | M | 31 | 174 | 289 | 89 | 96 | F508del | F508del | 0 | TP | |
| 61 | M | 31 | 186 | 158 | 97 | 103 | F508del | F508del | 0 | TP | |
| 62 | M | 34 | 127 | 55.80 | 16 | 21 | F508del | L206W | 1 | FN | Meconium plug |
| 63 | M | 24 | 180 | 149 | 105 | IS | c.489+1G>T | c.1679+1.6kbA>G | 0 | TP | |
| 64 | M | 180 | – | – | 97.4 | – | F508del | I507del | 0 | Screening not done due to parental decision | |
| 65 | M | 32 | 124 | 68 | 62 | 69 | R1066C | A457P | 1 | TP | |
| 66 | M | 31 | 76.5 | 59.70 | 44 | 49 | D1152H | F1052V | 1 | TP | |
| 67 | M | 300 | 56.6 | – | 59.6 | – | F508del | L206W | 1 | FN | Heat prostration |
| 68 | M | 48 | 116 | 109 | 96.3 | 99 | F508del | R1152H | 0 | TP | |
| 69 | M | 32 | 74.7 | 55.3 | 54.7 | 48.3 | R1066C | L206W | 1 | TP | |
| 70 | M | 37 | 71.1 | 45.5 | 25.6 | 29.3 | F508del | D1152H | 1 | FN | Genetics |
| 71 | M | 2 | 52.9 | – | 113.7 | – | F508del | F508del | 0 | FN | Meconium ileus |
| 72 | M | 985 | IS | 32.3 | 69.9 | 73 | F508del | L159S | 1 | FN | Growth impairment |
| 73 | M | 45 | 110 | 109 | 113 | 101.7 | F508del | c.2988+1G>A | 0 | TP |
CF: cystic fibrosis; CFNS: cystic fibrosis neonatal screening; F: female; FN: false negative; IRT: immunoreactive trypsinogen; IS: insufficient sample; M: male; PI: pancreatic insufficiency; PS: pancreatic sufficiency; TP: true positive.
Of these, 4347 (0.92%) had raised IRT1 and 1087 (0.23%) had raised IRT2. Since CFNS was introduced, 73 cases of CF were diagnosed; 60 were diagnosed by positive CFNS, and 13 were not diagnosed by CFNS. Specifically, of these 13 patients, 1 developed a typical clinical picture of CF, but was determined not to have undergone CFNS at the decision of the parents; the remaining 12 had negative CFNS (false negatives). Of these, 1 patient was diagnosed before symptoms appeared after his twin brother had a positive CFNS result. Another patient was diagnosed before symptoms appeared when chloride levels at the upper limit of normal were obtained and genetic studies were requested. The remaining 10 CFNS-negative patients were diagnosed when they developed clinical symptoms. Symptoms prompting the diagnosis of these 10 patients were: dehydration with hyponatremia, hypopotassemia, hypochloremia and metabolic alkalosis in 4 patients, a clinical picture of steatorrhea, growth impairment, and respiratory infections in 2, meconium ileus (MI) at birth in 2, meconium plug in 1, and growth impairment in 1. The characteristics of all patients diagnosed with CF since the implementation of the CFNS are listed in Table 1. In total, 46% were male and 54% were female. Mean age at diagnosis of the 60 patients diagnosed by CFNS was 39.86±12.50 days. Mean age at diagnosis of patients diagnosed from clinical signs and symptoms was 258.46±285.55 (2–985) days. Taking into account all data, sensitivity and specificity of the IRT1/IRT2 CFNS program was 83.33% and 99.78%, respectively, and the positive predictive value (PPV) and negative predictive value (NPV) were 5.52% and 99.99%, respectively. After excluding patients with clinical onset in the form of MI, sensitivity and specificity of the CFNS program in our community were 85.71% and 99.78%, respectively. The incidence of CF in Andalusia is 1:6506 live births. The 2 most frequently isolated mutations were F508del ([c.1521_1523delCTT] [75%]) and G542X ([c.1624G>T] [10.95%]). Table 2 summarizes the frequency of the different mutations.
Frequency of Mutations in Our Series.
| Mutation Type: Classic | DNA | Protein | No. of Patients (%) |
|---|---|---|---|
| F508del | c.1521_1523delCTT | p.Phe508del | 55 (75%); homozygous 20 (30.14%), heterozygous >35 (47.95%). |
| G542X | c.1624G>T | p.Gly542X | 8 (10.96%) |
| R334W | c.1000C>T | p.Arg334Trp | 7 (9.6%) |
| 2789+5G>A | c.2657+5G>A | 5 (6.8%) | |
| del I507 | c.1519_1521delATC | p.Ile507del | 4 (5.48%) |
| L206W | c.617T>G | p.Leu206Trp | 3 (4.11%) |
| N1303K | c.3909C>G | p.Asn1303Lys | 3 (4.11%) |
| D1152H | c.3454G>C | p.Asp1152His | 3 (4.11%) |
| R1066c | c.3196C>T | p.Arg1066Cys | 3 (4.11%) |
| S549R (T->G) | c.1647T>G | p.Ser549Arg | 2 (2.74%) |
| 1717-1G>A | c.1585-1G>A | – | 2 (2.74%) |
| G461R | c.1381G>A | p.Gly461Arg | 2 (2.74%) |
| W1282X | c.3846G>A | p.Trp1282X | 2 (2.74%) |
| R1162 | c.3484C>T | p.Arg1162X | 1 (1.37%) |
| Q1281X | c.3841C>T | p.Gln1281X | 1 (1.37%) |
| Q493X | c.1477C>T | p.Gln493X | 1 (1.37%) |
| IVS18-1G>A | 1 (1.37%) | ||
| G85E | 1 (1.37%) | ||
| L997F | c.2991G>C | p.Leu997Phe | 1 (1.37%) |
| 1677delTA | c.1545_1546delTA | p.Tyr515X | 1 (1.37%) |
| R117C | c.349C>T | p.Arg117Cys | 1 (1.37%) |
| 1609delCA | c.1477_1478delCA | p.Gln493ValfsX10 | 1 (1.37%) |
| 1 (1.37%) | |||
| c.2988+1KbDel8.6Kb | 1 (1.37%) | ||
| Q890X | c.2668C>T | p.Gln890X | 1 (1.37%) |
| 711+5G>A | c.579+5G>A | 1 (1.37%) | |
| Currently varies: 711+1G>T | c.579+1G>T | 1 (1.37%) | |
| 1717-8G>A | c.1585-8G>A | 1 (1.37%) | |
| S1196X | c.3587C>G | p.Ser1196X | 1 (1.37%) |
| 1819-1G>A | c.1680-1G>A | 1 (1.37%) | |
| 1812-1G>A | c.1680-1G>A | 1 (1.37%) | |
| 1811+1.6 kbA>G | c.1679+1.6kbA>G | 1 (1.37%) | |
| R1066C | c.3196C>T | p.Arg1066Cys | 1 (1.37%) |
| 1 (1.37%) | |||
| R1158X | c.3472C>T | p.Phe1052Val | 1 (1.37%) |
| L159S | c.476T>C | p.Leu159Ser | 1 (1.37%) |
| 3120+1G>A | c.2988+1G>A | 1 (1.37%) | |
| F1052V | c.3154T>G | p.Phe1052Val | 1 (1.37%) |
| A457P | p.Ala457Pro | p.1369G>A | (1.37%) |
Our study presents the first results from the CFNS program in our community, unknown to date. The program involves the double determination of IRT (IRT1/IRT2), which is shown to have a sensitivity of 85% and a specificity 99%, and a PPV and NPV of 5.70% and 99.99%, respectively. These rates are similar to those reported in other studies. Massie et al., for example, using the same protocol reported 86.6% sensitivity, 99.4% specificity, and PPV and NPV of 3.5% and 99.9%, respectively.10 The PPV of the IRT/IRT program is 5%–9% according to some authors.4,11,12 The results of this study based on the IRT1/IRT2 protocol are similar to those obtained by other international CFNS centers, and can therefore be extrapolated to countries or regions using the same CFNS program. Thus, in the state of São Paulo (Brazil), investigators reported a sensitivity of up to 87%, a false positive rate of 95.2% and a PPV of 8%13; in the region of Lazio (Italy), sensitivity from 1992 to 1997 was 83%14; in the region of Lombardy (Italy) between 1990 and 1992, sensitivity was 84.7%, specificity 98.6%, and the rate of false negatives was 15%15; Poland introduced the IRT1/IRT2 protocol between January 1999 and June 2000 and obtained a mean sensitivity of 92.31% (74.87%–99.05%), specificity of 99.80%, and a PPV of 7.64% (4.96%–11.16%)16; between 1989 and 1990 in Victoria (Australia), sensitivity was 86.6%, specificity 99.4%, PPV 3.5%, and NPV 99.9%.10
Although we know that no single CFNS program will detect all cases of CF,4 sensitivity can be increased by introducing a panel of mutations, which must be in line with the genetics of the screened population,4 underlining the importance of including mutations found in confirmed clinical cases in the population examined.4 Massie et al. reported 89.9% sensitivity after determination of the F508del, but this figure rose to 95.8% when the DNA panel was expanded to 12 mutations.10 The inclusion of sequencing (NGS) in the CFNS algorithm has been shown to increase sensitivity, specificity, and PPV.2 At present, it seems that a multilevel approach using IRT, analysis of a basic panel of mutations, and finally CFTR sequencing is the most appropriate methodology for CFNS,2 although the cost and response time for results would probably have an impact on the suitability of this screening algorithm.2 DNA analysis detects not only conventional forms of CF, but also mutations of uncertain significance, and carriers of CF mutations.2,9,17 There is disagreement surrounding the identification of CF carriers due to the possible psychological impact,2,9,17 but we believe that, with genetic counselling, this strategy can contribute to reducing the incidence of the disease. A new marker called PAP has been used in recent years to obviate the need to identify CFTR mutations.2,9,17 Duplicate quantification of PAP and IRT in dry blood samples in the first week of life improves the specificity of CFNS programs, reducing the number of newborns who required sweat and/or genetic testing, thus reducing associated family anxiety.9 Studies have shown that the IRT/PAP strategy might be viable in terms of cost per case detected, although the detection rates of classic forms of CF may be lower than with IRT/DNA strategies.17 According to Seror et al., the detection rate of classic forms of CF obtained with the IRT/PAP strategy was similar to that of the IRT/DNA and IRT/PAP/DNA approaches, but the IRT/PAP strategy requires fewer resources and led to the detection of a lower number of mild forms compared to IRT/DNA.17,18 Switching from an IRT/DNA strategy to a IRT/PAP strategy would mean missing the opportunity to identify CF carriers.17Table 3 shows a comparison of the results of different CFNS programs.
Comparison of the Results of the Various CFNS Protocols.
| Our Protocol | IRT/IRT | IRT/p.508del | IRT/4 Mutations | IRT/12 Mutations | IRT/PAP (Cutoff ≥1 ng/ml) | IRT/PAP (Suggested Cutoff >0.9 ng/ml) | IRT/DNA/IRT | IRT/PAP/DNA | |
|---|---|---|---|---|---|---|---|---|---|
| Sensitivity | 85.71% | 86.6% | 89.9% | 71.4% | 95.8% | 85.7% | 92.8% | 90.48% | 71.4% |
| Specificity | 99.78% | 99.4% | 99.9% | 99.9% | 99.9% | 99.9% | 99.9% | 99.91% | 99.9% |
| Positive predictive value | 5.7% | 3.5% | 20.1% | 17.9% | 18.3% | 12.2% | 11.6 | 16.10 | 62.5% |
| Negative predictive value | 99.9% | 99.9% | 99.9% | 99.9% | 99.9% | 99.9% | 99.9% |
Early diagnosis of a CF patient is very beneficial.2,3,19 Early diagnosis and treatment of CF improves the infant's nutritional status, growth curve, cognitive development, and lung function, and reduces chronic colonizations, exacerbations, antibiotic requirements, and hospitalizations.2–4,19 The literature suggests that CF patients diagnosed within the first 2 months of life are more likely to benefit from early interventions.4,20 Implementation of our CFNS program allowed us to diagnose 79.45% of CF patients before the age of 8 weeks, with a mean age at diagnosis of only 39.86 days, compared to the mean delay of 258 days in patients who were not diagnosed with the CFNS protocol. This percentage is slightly higher than that found by other authors such as Kharrazi et al. who diagnosed 74.5% of newborns with CF before the age of 2 months.4
Among the false negatives, the presence of low levels of IRT in 2 patients (16.67%) with MI should be noted (a finding also reported by other authors1,2,21), despite both patients having severe mutations, namely homozygous F508del in one and heterozygous p.lle507 in the other. Four patients with MI in our series were diagnosed using CFNS, 3 of which were homozygous carriers of the F508del mutation. Newborns with MI have a higher risk of a false negative result on CFNS than newborns without MI, since in some cases, the IRT levels of CF patients with MI are lower than in CF patients without MI.2 For this reason, as in some other countries,2 we emphasize the importance of reporting the presence of MI as part of the CFNS program, so that a sweat test can be performed in these patients, if feasible according to their clinical status, and/or a genetic study for CF.
The most common mutation in our population was F508del (c.1521_1523delCTT), found in 75% of patients, a higher rate than that of other authors who reported that this mutation accounts for approximately 60%–70% of the mutations causing CF in Caucasian populations of European origin.1–3 The most commonly isolated mutations in other countries such as the United Kingdom include F508del (74.1%), G551D [(c.1652G>A)(3.37%)] and G542X (1.85%)3; in Denmark up to 88% of cases have F508del compared to 50% in Italy and 20% in Turkey. In the Balearic Islands, the rate is 58.5%, similar to Galicia, while in the Basque Country, it is as high as 87%. The second most common mutation was G542X, that occurred at a rate of 10.95%, similar to that found in the Balearic Islands, and significantly higher than in Asturias (5%), Castile-Leon (3.8%), and Galicia (4.9%).3 L206W (c.617T>G) was isolated in 4.1% of our cases, a rate higher than the overall rate in Spain (1.6%). This mutation is more prevalent in the Hispanic population (6.2%) and less so among African Americans (1.2%).3
With this study, we have been able to determine for the first time that the incidence of CF in Andalusia is 1:6506. According to data from the Spanish Association of Neonatal Screening (AECNE), the overall incidence in Spain is 1:4807,3 although differences have been observed between the various autonomous communities.3 Thus, in Galicia and Castile-Leon it is 1:4439, 1:4800 in Aragon, 1:6244 in Catalonia, and 1:6602 in the Balearic Islands.3,22 We anticipate that the implementation of the CFNS program and the subsequent genetic counseling will decrease the incidence of the disease in our community, as has occurred in other regions.23
One of the main strengths of this study is the high level of compliance with the CFNS program, which offers a good representation of the population of Andalusia. Despite the fact that our observation period was relatively short, these results allow us to reflect on possible areas for further improvement of the CFNS algorithm, which should include the introduction of a genetic study in our program in order to improve sensitivity, reduce the rate of false positives, and mitigate family anxiety.
Conflict of InterestsThe authors state that they have no conflict of interests.
Please cite this article as: Delgado Pecellín I, Pérez Ruiz E, Álvarez Ríos AI, Delgado Pecellín C, Yahyaoui Macías R, Carrasco Hernández L, et al. Resultados del programa de screening neonatal de fibrosis quística en Andalucía tras 5 años de su implantación. Arch Bronconeumol. 2018;54:551–558.







