






Andalucía dispone de screening neonatal de fibrosis quística (SNFQ) desde mayo 2011, basado en doble determinación de tripsinógeno inmunorreactivo ([TIR] [TIR1/TIR2]). Si el screening es positivo realizamos un test del sudor y si es positivo o dudoso solicitamos genética.
ObjetivoAnalizar el SNFQ, basado en los resultados de los primeros 4,5 años.
Material y métodoEstudio descriptivo prospectivo de los neonatos sometidos a SNFQ. Se recogen los niveles de TIR, cloruro en sudor, mutaciones. Mediante SPSS12.0 se realizó análisis estadístico.
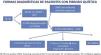
ResultadosDesde mayo 2011 a diciembre 2016, 474.953 neonatos fueron sometidos a SNFQ. Mil ochenta y siete (0,23%) presentaron TIR2 elevado. Desde la implantación del SNFQ se diagnosticaron 73 casos con fibrosis quística; 60 de ellos fueron diagnosticados mediante un SNFQ positivo, mientras que 13 no. Concretamente un paciente comenzó con clínica clásica de fibrosis quística y se comprobó que no se había realizado el SNFQ por decisión paterna; los 12 restantes tuvieron un SNFQ negativo (falsos negativos). De estos, un paciente fue diagnosticado presintomáticamente al tener su hermano gemelo con SNFQ positivo; otro con cloruro en el límite alto de la normalidad se diagnosticó presintomáticamente mediante genética; 10 pacientes comenzaron clínicamente. Excluyendo los pacientes con íleo meconial, la sensibilidad y especificidad del programa de SNFQ asciende al 85,71 y 99,78% respectivamente. La incidencia de la enfermedad en Andalucía es de 1/6.506 recién nacidos vivos.
ConclusiónLos presentes resultados nos permiten reflexionar sobre posibles áreas de mejoras adicionales del algoritmo del SNFQ, que debe pasar por la introducción de estudios genéticos para así aumentar la sensibilidad y disminuir los falsos positivos.
Cystic fibrosis neonatal screening (CFNS), based on double determination of immunoreactive trypsinogen ([IRT] [IRT1/IRT2]), has been available in Andalusia since May 2011. If screening is positive, a sweat test is performed, and if that is positive or inconclusive, genetic testing is requested.
ObjectiveTo analyze CFNS, based on results from the first 4.5 years of the program.
Materials and methodsProspective descriptive study of neonates undergoing CFNS. IRT levels, sweat chloride, and mutations were recorded. Statistical analysis was performed using SPSS 12.0.
ResultsBetween May 2011 and December 2016, 474,953 neonates underwent CFNS. Of these, 1,087 (0.23%) had elevated IRT2. Since CFNS was introduced, 73 cases of cystic fibrosis were diagnosed; 60 were diagnosed by positive CFNS, and 13 were diagnosed by other means. In one case, the patient developed a typical clinical picture of cystic fibrosis, but had not undergone CFNS at the decision of the parents; the remaining 12 had a negative CFNS (false negatives). Of these, one patient was diagnosed before symptoms developed, as his twin brother had a positive CFNS result; another had chloride at the upper limit of normal, and was subsequently diagnosed with genetic testing before symptoms appeared; and 10 patients developed clinical signs and symptoms. Excluding patients with meconium ileus, sensitivity and specificity of the CFNS program were 85.71% and 99.78%, respectively. The incidence of the disease in Andalusia is 1/6,506 live births.
ConclusionThese results are a basis for reflection on possible areas for improvement of the CFNS algorithm, and thought may be given to the introduction of genetic studies to increase sensitivity and reduce false positives.
La fibrosis quística (FQ, OMIM # 219700) es una enfermedad crónica, multisistémica, que en la mayoría de los casos asocia infecciones respiratorias, insuficiencia pancreática y aumento de la concentración de cloruro en el sudor debido a mutaciones en el gen regulador de la conductancia transmembrana (CFTR)1–3. Hasta la fecha se han identificado más de 2.000 variantes genéticas1,2,4. En la actualidad estamos asistiendo a un importante aumento de la supervivencia gracias a mejoras en la nutrición, mayor acceso a la antibioterapia sistémica e inhalada, al uso de enzimas pancreáticas, mejoras en el trasplante pulmonar, al tratamiento con moduladores de la proteína CFTR o a la implantación de programas de cribado neonatal1,2,5–7.
Actualmente no existe un programa universalmente aceptado de screening neonatal de FQ (SNFQ), pero todos utilizan como primer marcador la cuantificación de tripsinógeno inmunorreactivo (TIR)3,8,9. Los niveles de TIR están elevados en la mayoría de los pacientes con FQ, pero también pueden aumentar por otros motivos, incluidos prematuridad y bajo peso al nacer, constituyendo causas de falsos positivos y de elevada ansiedad familiar1,2,8. Para aumentar la especificidad (E), generalmente se determina un segundo marcador que puede ser la proteína asociada a pancreatitis (PAP), una segunda cuantificación de TIR (TIR2) o determinación de mutaciones de CFTR1,3,8,9.
Hasta el momento actual no disponemos de datos sobre el protocolo más idóneo para realizar este cribado. Conocer los resultados de programas de cribado actualmente implementados es un paso necesario para conocer la rentabilidad de los distintos algoritmos que permitan en un futuro desarrollar una estrategia consensuada.
En Andalucía, el SNFQ se introdujo en mayo de 2011, como parte del cribado neonatal ampliado de 30 enfermedades. El objetivo de este estudio fue evaluar los resultados del programa andaluz de SNFQ, basado en los resultados de los primeros 4,5 años, describir sus fortalezas, identificar las posibilidades de mejoras y comparar o extrapolar los resultados a otras comunidades o países con el mismo protocolo de SNFQ.
Material y métodosEstudio observacional de diseño transversal en el que incluyeron todos los recién nacidos en Andalucía entre el 1 de mayo de 2011 y el 31 de diciembre de 2016. El screening neonatal está centralizado en 2 centros: el Hospital Universitario Virgen del Rocío de Sevilla y el Hospital Regional Universitario Carlos Haya de Málaga. Las muestras de sangre se recogen, tras punción en el talón, en papel de filtro con dispositivos de recolección de muestras Perkin Elmer 226 (Waltham, Massachusetts, EE. UU.) a las 48-72 h de vida del neonato de vida del neonato. Estas muestras de sangre seca se enviaron por correo a las 2 unidades de referencia de cribado neonatal en Andalucía según una distribución geográfica. Estas muestras se analizan mediante inmunoanálisis usando el kit AutoDELFIA Neonatal IRT para la determinación de TIR.
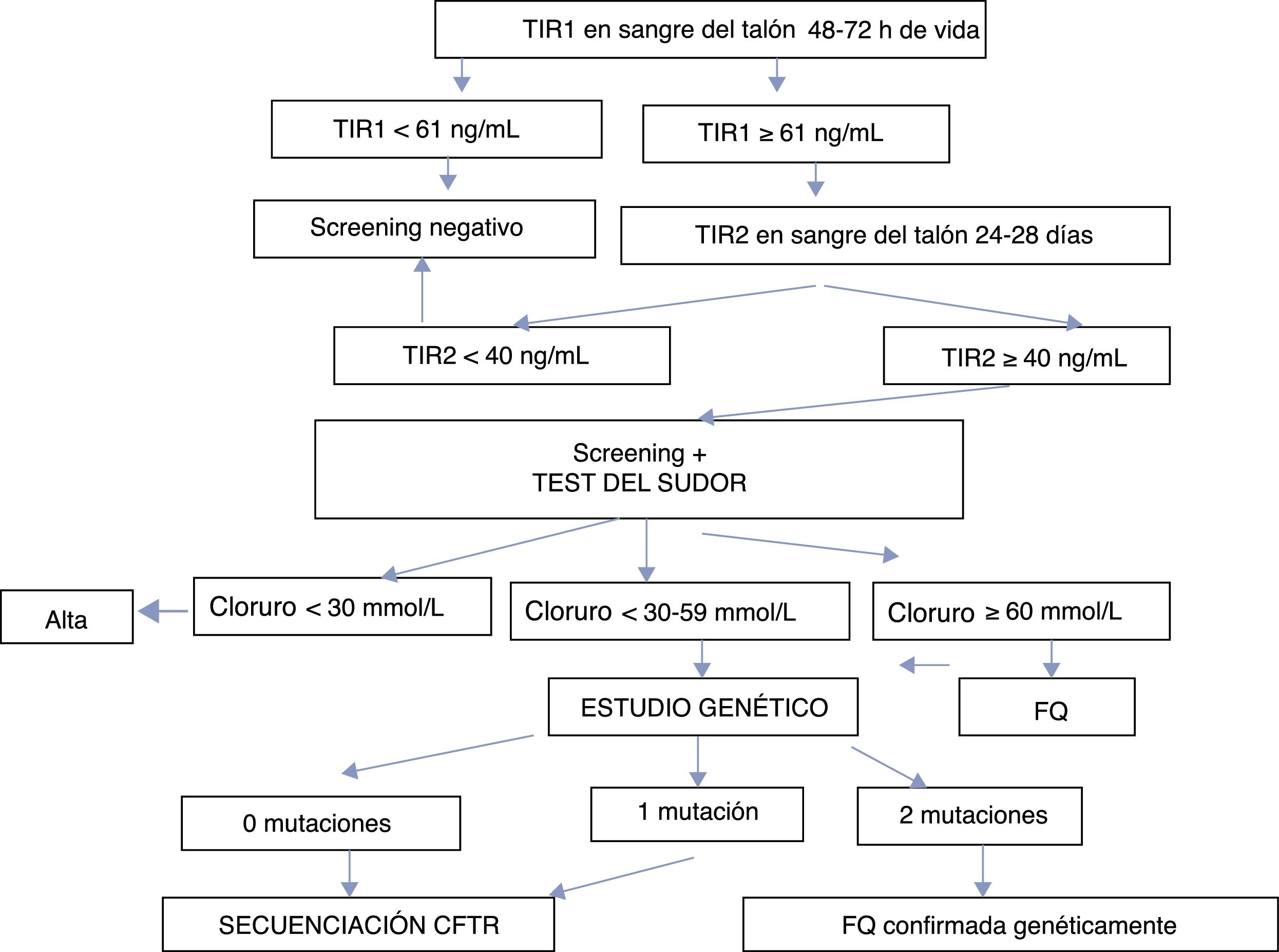
El programa de cribado está basado en la doble determinación de TIR. La primera TIR (TIR1) se cuantifica a las 48-72h de vida. Si alcanza valores≥61ng/mL, se solicita una segunda determinación entre los días 25-28 de vida del neonato. Si TIR2 es≥40ng/mL, se remite al neonato a una unidad de referencia para realización del test del sudor. En caso de tener un test de sudor dudoso o positivo (cloruro≥30mmol/L), se realiza estudio genético. La figura 1 resume el programa de SNFQ en Andalucía.
Las pruebas de sudor se realizaron en al menos 2 ocasiones mediante iontoforesis con pilocarpina utilizando un dispositivo de recolección Macroduct® (Wescor, Logan, UT. EE. UU.) con mediciones de cloruro en un MK II Chloride Analyzer 926S (Sherwood Scientific, Ltd).
Se realizaron pruebas genéticas confirmatorias para validar los resultados de TIR y del test del sudor. Primero se procedió a la extracción automática de ADN del paciente a partir de una muestra de sangre periférica (MagNA Pure Compact, Roche, Alemania). En un primer paso se realizó el Kit Elucigene CF-EU2 (Elucigene Diagnostics, Reino Unido) que permite la detección de 50 mutaciones de forma simultánea utilizando la tecnología ARMS (Amplification Refractory Mutation System). Los fragmentos generados fueron analizados mediante electroforesis capilar en el secuenciador ABI3730 empleado el software GeneMapper v 4.0 (ThermoFisher Scientific, Waltham, Massachusetts, EE. UU.). En los casos con test del sudor dudoso o positivo en los que mediante el panel de 50 mutaciones no se identificaron las 2 mutaciones, se realizó secuenciación completa del gen CFTR mediante secuenciación masiva, utilizando el kit CFTR MASTRTM Dx de Multiplicom y el secuenciador MiSeq® (lllumina). El análisis de los datos de secuenciación masiva se llevó a cabo utilizando el módulo SeqNext del Software Sequence Pilot de JSI medical systems.
El estudio cumplió con los requisitos éticos de la Declaración de Helsinki sobre estudios con seres humanos y con el reglamento español sobre protección de datos y confidencialidad (Ley Orgánica 15/1999, de 13 de diciembre, de Protección de Datos Personales). Los registros clínicos se anonimizaron en la base de datos mediante la asignación de un código numérico a través de un algoritmo. No se ingresó información personal que pudiera usarse directa o indirectamente para identificar a un individuo. La relación entre el código de la base de datos y el número de historia clínica se mantuvo localmente y era responsabilidad del investigador local. En todos los casos con estudio genético, se solicitó consentimiento informado a los padres por escrito.
Análisis estadísticoEl análisis estadístico se realizó con el software SPSS12.0. Tras comprobar el resultado del cribado y del diagnóstico genético definitivo, se evaluó el perfil diagnóstico calculando la sensibilidad (S), E y valores predictivos con sus intervalos de confianza. El valor de significación se estableció en 0,05.
ResultadosDesde el 1 de mayo de 2011 al 31 de diciembre de 2016, 474.953 recién nacidos fueron sometidos a SNFQ evaluándose el TIR1 usando el sistema AutoDELFIA. Una visión general del algoritmo diagnóstico y los resultados se presentan en la figura 1 y tabla 1 respectivamente. Los análisis de tendencia de los percentiles se revisaron mensualmente. El límite de TIR1 se estableció inicialmente en 65ng/mL y de TIR2 en 35mmol/L, hasta noviembre de 2012, momento en el que se ajustaron a 60 y 40ng/mL re
Resumen de las características de todos los pacientes diagnosticados de FQ desde la implantación del SNFQ
| Sexo | Días al diagnóstico | TIR1 (ng/ml) | TIR2 (ng/ml) | 1.o cloro en sudor (mmol/l) | 2.o cloro en sudor (mmol/l) | Mutación 1 | Mutación 2 | Suficiencia pancreática. (0=IP;1=SP) | Diagnóstico | Síntomas diagnósticos de los FN. Observaciones | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | V | 15 | 112 | -- | 103 | MI | F508del | F508del | 0 | VP | Íleo meconial |
| 2 | V | 600 | -- | -- | 55 | 60 | c.2657+5G>A | Desconocida | 1 | FN | Postración por calor |
| 3 | M | 87 | 120 | 59 | 42 | MI | F508del | D1152H | 1 | VP | |
| 4 | V | 52 | 115 | 69 | 79 | MI | R334W | Q1281X | 1 | VP | |
| 5 | V | 34 | 218 | 369 | 103 | MI | F508del | IVS18-1 G>A | 0 | VP | |
| 6 | V | 51 | 140 | 73 | 81 | MI | F508del | G461R | 1 | VP | |
| 7 | M | 42 | 151 | 114 | 60 | 56 | G542X | G461R | 1 | VP | |
| 8 | V | 35 | 86 | 123 | 81 | MI | F508del | G542X | 0 | VP | |
| 9 | V | 34 | 182 | 152 | 100 | MI | F508del | I507del | 0 | VP | |
| 10 | V | 34 | 139 | 128 | 65 | MI | F508del | F508 | 0 | VP | |
| 11 | M | 40 | 112 | 149 | 93 | MI | G85E | Q493X | 1 | VP | |
| 12 | V | 600 | MI | 7 | 112 | MI | p.Leu997Phe | c.2909-92G>A | 0 | FN | Clínica clásica |
| 13 | V | 43 | 194 | 92 | 73 | MI | F508del | F508del | 0 | VP | |
| 14 | V | 31 | 153 | 144 | 85 | MI | F508del | G542X | 0 | VP | |
| 15 | M | 43 | 92 | 175 | 115 | MI | F508del | p.Tyr515X | 0 | VP | |
| 16 | V | 15 | 331 | -- | 94 | MI | F508del | F508del | 0 | VP | Íleo meconial |
| 17 | V | 146 | 89 | 34 | 87 | MI | F508del | F508del | 0 | FN | Clínica clásica |
| 18 | V | 60 | 97 | 161 | 110 | MI | F508del | F508del | 0 | VP | |
| 19 | V | 34 | 61 | 125 | 90 | MI | F508del | F508del | 0 | VP | |
| 20 | M | 240 | 48 | -- | 73 | MI | N1303K | R117C | 1 | FN | Deshidratación |
| 21 | V | 25 | 62 | 107 | 82 | MI | F508del | F508del | 0 | VP | Íleo meconial |
| 22 | M | 36 | 187 | 60 | 101 | MI | F508del | W1282X | 0 | VP | |
| 23 | M | 41 | 126 | 386 | -- | MI | F508del | p.Glyn493ValfsX10 | 0 | VP | |
| 24 | M | 48 | 66 | 77 | 77 | MI | p.Arg1162X | Desconocida | 1 | VP | |
| 25 | V | 32 | 122 | 160 | 90 | MI | F508del | F508del | 0 | VP | |
| 26 | V | 30 | 232 | 121 | 84 | MI | I507del | Desconocida | 0 | VP | |
| 27 | V | 40 | 96 | 88 | MI | MI | F508del | W1282X | 0 | VP | |
| 28 | M | 65 | 200 | 310 | 79 | MI | c.579+5G>A | c.2988+1KbDel8.6Kb | 0 | VP | |
| 29 | V | 19 | 85 | 145 | 90 | MI | c.579+1G>T | Q890X | 0 | VP | Íleo meconial |
| 30 | M | 58 | 263 | 300 | MI | MI | F508del | c.1585-1G>A | 0 | VP | |
| 31 | M | 58 | 161 | 254 | MI | MI | F508del | c.1585-1G>A | 0 | VP | |
| 32 | M | 61 | 140 | 178 | MI | MI | F508del | S549R | 0 | VP | |
| 33 | V | 51 | 147 | 211 | 116 | MI | F508del | c.1585-85>A | 0 | VP | |
| 34 | M | 59 | 103 | -- | 120 | 93,7 | F508del | G542X | 0 | VP | |
| 35 | M | 58 | 63,6 | 44,6 | 119 | 106,1 | N1303K | R334W | 1 | VP | |
| 36 | M | 58 | 61,4 | 41,3 | 113 | 101,8 | N1303K | R334W | 1 | VP | |
| 37 | V | 42 | 75,6 | 63 | 99 | 76 | F508del | c.2657+5G>A | 1 | VP | |
| 38 | V | 42 | 39,3 | -- | 98 | 72,5 | F508del | c.2657+5G>A | 1 | FN | Hermano gemelo con screening + |
| 39 | M | 34 | 133 | 113 | 122 | 92,9 | R334w | S549R | 1 | VP | |
| 40 | V | 35 | 85,6 | 115 | 102 | 109 | F508del | G542X | 0 | VP | |
| 41 | V | 32 | 101 | 79 | -- | -- | F508del | R334W | 1 | VP | |
| 42 | M | 35 | 148 | 175 | 112 | MI | F508del | G542X | 0 | VP | |
| 43 | M | 33 | 93,7 | 233 | 100 | 77,1 | F508del | F508del | 0 | VP | |
| 44 | V | 36 | 58,4 | 52,4 | 78 | 81 | F508del | R334W | 1 | VP | |
| 45 | V | 34 | 212 | 165 | 101 | 98 | F508del | S1196X | 0 | VP | |
| 46 | M | 45 | 230 | 496 | 109 | 107 | F508del | F508del | 0 | VP | |
| 47 | V | 180 | 33 | -- | 59 | 57 | F508del | c.3717+10kbC>T | 1 | FN | Postración por calor |
| 48 | M | 36 | 90,7 | 45,70 | 89 | 96 | F508del | R334W | 1 | VP | |
| 49 | M | 34 | 176 | 70,30 | 101 | 110 | F508del | F508del | 0 | VP | |
| 50 | M | 14 | 41 | -- | 74 | -- | p.Ile507del | F508del | 0 | FN | Íleo meconial |
| 51 | V | 36 | 105 | 70,90 | 29 | 32 | F508del | c.2657+5G>A | 1 | VP | |
| 52 | V | 36 | 94,9 | 62,10 | 64 | 69 | F508del | c.2657+5G>A | 1 | VP | |
| 53 | V | 32 | 132 | 210 | 105 | 79,4 | F508del | F508del | 0 | VP | |
| 54 | V | 36 | 120 | 448 | 112 | 94,7 | F508del | F508del | 0 | VP | |
| 55 | V | 32 | 132 | 422 | 87 | MI | F508del | F508del | 0 | VP | |
| 56 | M | 35 | 239 | 287 | 118 | 103 | F508del | F508del | 0 | VP | |
| 57 | M | 35 | 151 | 179 | 104 | 98 | F508del | G542X | 0 | VP | |
| 58 | V | 33 | 169 | 169 | 100 | 91,1 | F508del | F508del | 0 | VP | |
| 59 | M | 36 | 122 | 82,90 | 105 | 101 | c.1680-1G>A | G542X | 0 | VP | |
| 60 | M | 31 | 174 | 289 | 89 | 96 | F508del | F508del | 0 | VP | |
| 61 | M | 31 | 186 | 158 | 97 | 103 | F508del | F508del | 0 | VP | |
| 62 | V | 34 | 127 | 55,80 | 16 | 21 | F508del | L206W | 1 | FN | Tapón meconial |
| 63 | M | 24 | 180 | 149 | 105 | MI | c.489+1G>T | c.1679+1.6kbA>G | 0 | VP | |
| 64 | V | 180 | -- | -- | 97,4 | -- | F508del | I507del | 0 | Screening no realizado por decisión paterna | |
| 65 | V | 32 | 124 | 68 | 62 | 69 | R1066C | A457P | 1 | VP | |
| 66 | V | 31 | 76,5 | 59,70 | 44 | 49 | D1152H | F1052V | 1 | VP | |
| 67 | M | 300 | 56,6 | -- | 59,6 | -- | F508del | L206W | 1 | FN | Postración por calor |
| 68 | M | 48 | 116 | 109 | 96,3 | 99 | F508del | R1152H | 0 | VP | |
| 69 | M | 32 | 74,7 | 55,3 | 54,7 | 48,3 | R1066C | L206W | 1 | VP | |
| 70 | M | 37 | 71,1 | 45,5 | 25,6 | 29,3 | F508del | D1152H | 1 | FN | Genética |
| 71 | M | 2 | 52,9 | -- | 113,7 | -- | F508del | F508del | 0 | FN | Íleo meconial |
| 72 | V | 985 | MI | 32,3 | 69,9 | 73 | F508del | L159S | 1 | FN | Fallo de medro |
| 73 | V | 45 | 110 | 109 | 113 | 101,7 | F508del | c.2988+1G>A | 0 | VP |
FN: falsos negativos; FQ: fibrosis quística; IP: insuficiencia pancreática; M: mujer; MI: muestra insuficiente; SNFQ: screening neonatal de fibrosis quística; SP: suficiencia pancreática; TIR: tripsinógeno inmunorreactivo; V: varón; VP: verdadero positivo.
spectivamente, representando el percentil 99,5 de TIR acumulado en ese punto.
De ellos 4.347 (0,92%) tuvieron un TIR1 elevado y 1.087 (0,23%) presentaron un TIR2 elevado. Desde la implantación del SNFQ se diagnosticaron un total de 73 niños con FQ; 60 de ellos fueron diagnosticados mediante un SNFQ positivo, mientras que 13 no fueron diagnosticados mediante el SNFQ. Concretamente, de esos 13 pacientes, uno comenzó con clínica clásica de FQ y se comprobó que no se había realizado el SNFQ por decisión de los padres, mientras que los 12 restantes tuvieron un SNFQ negativo (falsos negativos). De estos, un paciente fue diagnosticado presintomáticamente al tener un hermano gemelo con SNFQ positivo, otro paciente se diagnosticó presintomáticamente tras obtener niveles de cloro en el límite alto de la normalidad y solicitar genética y los 10 pacientes restantes con SNFQ negativo comenzaron clínicamente. Los síntomas al diagnóstico de esos 10 pacientes fueron: deshidratación hiponatrémica, hipopotasémica e hipoclorémica con alcalosis metabólica en 4 pacientes, 2 presentaron clínica clásica consistente en esteatorrea, fallo de medro e infecciones respiratorias, 2 pacientes comenzaron con íleo meconial (IM) al nacer, uno con tapón meconial y otro con fallo de medro. La tabla 1 resume las características de todos los pacientes diagnosticados de FQ desde la implantación del SNFQ. El 46% fueron varones frente al 54% que fueron niñas. La edad media al diagnóstico de los 60 pacientes diagnosticados mediante SNFQ fue de 39,86±12,50 días. La edad media al diagnóstico de los pacientes diagnosticados mediante clínica fue 258,46±285,55 (2-985) días. Teniendo en cuenta todos los datos, la S y la E del programa de SNFQ TIR1/TIR2 han sido del 83,33% y del 99,78 respectivamente y el valor predictivo positivo (VPP) y valor predictivo negativo (VPN) del 5,52% y del 99,99% respectivamente. Excluyendo los pacientes que han comenzado clínicamente con un IM, la S y E del programa de SNFQ en nuestra comunidad asciende a 85,71% y 99,78% respectivamente. La incidencia de la enfermedad en Andalucía es de 1 paciente con FQ por cada 6.506 recién nacidos vivos. Las 2 mutaciones más frecuentemente aisladas han sido F508del ([c.1521_1523delCTT] [75%]) y G542X ([c.1624G>T] [10,95%]). La tabla 2 resume la frecuencia de las diferentes mutaciones.
Frecuencia de las mutaciones en nuestra población
| Tipo mutación: clásica | ADN | Proteína | N.o pacientes (%) |
|---|---|---|---|
| F508del | c.1521_1523delCTT | p.Phe508del | 55 (75%); homocigotos 20 (30,14%); heterocigotos 35 (47,95%) |
| G542X | c.1624G>T | p.Gly542X | 8 (10,96%) |
| R334W | c.1000C>T | p.Arg334Trp | 7 (9,6%) |
| 2789+5G>A | c.2657 + 5G>A | 5 (6,8%) | |
| del I507 | c.1519_1521delATC | p.Ile507del | 4 (5,48%) |
| L206W | c.617T>G | p.Leu206Trp | 3 (4,11%) |
| N1303K | c.3909C>G | p.Asn1303Lys | 3 (4,11%) |
| D1152H | c.3454G>C | p.Asp1152His | 3 (4,11%) |
| R1066c | c.3196C>T | p.Arg1066Cys | 3 (4,11%) |
| S549R (T->G) | c.1647T>G | p.Ser549Arg | 2 (2,74%) |
| 1717-1G>A | c.1585-1G>A | --- | 2 (2,74%) |
| G461R | c.1381G>A | p.Gly461Arg | 2 (2,74%) |
| W1282X | c.3846G>A | p.Trp1282X | 2 (2,74%) |
| R1162 | c.3484C>T | p.Arg1162X | 1 (1,37%) |
| Q1281X | c.3841C>T | p.Gln1281X | 1 (1,37%) |
| Q493X | c.1477C>T | p.Gln493X | 1 (1,37%) |
| IVS18-1G>A | 1 (1,37%) | ||
| G85E | 1 (1,37%) | ||
| L997F | c.2991 G>C | p.Leu997Phe | 1 (1,37%) |
| 1677delTA | c.1545_1546delTA | p.Tyr515X | 1 (1,37%) |
| R117C | c.349C>T | p.Arg117Cys | 1 (1,37%) |
| 1609delCA | c.1477_1478delCA | p.Gln493ValfsX10 | 1 (1,37%) |
| 1 (1,37%) | |||
| c.2988+1KbDel8.6Kb | 1 (1,37%) | ||
| Q890X | c.2668C>T | p.Gln890X | 1 (1,37%) |
| 711+5G>A | c.579+5G>A | 1 (1,37%) | |
| actualmente varía: 711+1G>T | c.579+1G>T | 1 (1,37%) | |
| 1717-8G>A | c.1585-8G>A | 1 (1,37%) | |
| S1196X | c.3587C>G | p.Ser1196X | 1 (1,37%) |
| 1819-1G>A | c.1680-1G>A | 1 (1,37%) | |
| 1812-1G>A | c.1680-1G>A | 1 (1,37%) | |
| 1811+1.6 kbA>G | c.1679 + 1.6kbA>G | 1 (1,37%) | |
| R1066C | c.3196C>T | p.Arg1066Cys | 1 (1,37%) |
| 1 (1,37%) | |||
| R1158X | c.3472C>T | p.Phe1052Val | 1 (1,37%) |
| L159S | c.476T>C | p.Leu159Ser | 1 (1,37%) |
| 3120+1G>A | c.2988+1G>A | 1 (1,37%) | |
| F1052V | c.3154T>G | p.Phe1052Val | 1 (1,37%) |
| A457P | p.Ala457Pro | p.1369G>A | (1,37%) |
Nuestro estudio presenta por primera vez los resultados del programa de SNFQ existente en nuestra comunidad, desconocidos hasta la fecha. El programa implantado consiste en la doble determinación de TIR (TIR1/TIR2), mostrando una S alrededor del 85% y una E del 99%, un VPP y VPN del 5,70% y del 99,99% respectivamente, datos similares a los publicados por otros autores. Así, Massie et al. han publicado una S del mismo programa de SNFQ del 86,6%, una E del 99,4% y un VPP y VPN del 3,5% y del 99,9% respectivamente10. El VPP del programa TIR/TIR es del 5-9% según algunos autores4,11,12. Los resultados del presente estudio basado en el protocolo TIR1/TIR2 son similares a los obtenidos por otros centros internacionales de SNFQ y por tanto extrapolables a aquellos países o regiones con el mismo programa de SNFQ. Así, en estado de San Pablo (Brasil) obtuvieron una S de hasta el 87%, una tasa de falsos positivos del 95,2% y un VPP del 8%13; en la región de Lacio (Italia) la S encontrada de 1992 a 1997 fue del 83%14; en la región de Lombardía (Italia) entre los años 1990-1992 encontraron una S del 84,7%, E del 98,6% y una tasa del falsos negativos del 15%15; Polonia implantó entre enero 1999 y junio 2000 el protocolo TIR1/TIR2, obteniendo una S media del 92,31% (74,87-99,05%), una E del 99,80% y un VPP del 7,64% (4,96-11,16%)16; en Victoria (Australia) entre 1989-1990 obtuvieron una S del 86,6%, una E de 99,4%, VPP del 3,5% y un VPN del 99,9%10.
Aunque sabemos que ningún programa de SNFQ detectará todos los casos de FQ4, es posible aumentar la S mediante la introducción de un panel de mutaciones, que debe ser acorde a la genética de la población cribada4, destacando la importancia de incluir mutaciones encontradas en casos clínicos confirmados en la población examinada4. Así Massie et al. encuentran una S del 89,9% tras la determinación de la mutación F508del, pero que asciende hasta el 95,8% cuando amplía el panel de ADN a 12 mutaciones10. Se ha demostrado que la incorporación de la secuenciación (NGS) dentro del algoritmo de SNFQ aumenta la S, la E y el VPP2. En el momento actual, parece que un enfoque de múltiples niveles que utiliza TIR, el análisis de un panel básico de mutaciones y, finalmente, la secuencia del gen CFTR sería la metodología más adecuada para el SNFQ2, aunque el coste y el tiempo de respuesta a los resultados probablemente influirán en la idoneidad del algoritmo del mencionado cribado2. El análisis de ADN detecta tanto formas clásicas como leves de FQ, mutaciones de significado incierto, así como portadores de mutaciones de FQ2,9,17. Existe algún desacuerdo sobre los posibles efectos ocasionados por la identificación de portadores de FQ debido a los posibles efectos psicológicos2,9,17, aunque pensamos que su identificación puede contribuir a reducir la incidencia de la enfermedad, tras un consejo genético. Para evitar la determinación de mutaciones de CFTR en los últimos años se está utilizando un nuevo marcador denominado PAP2,9,17. La cuantificación de PAP y TIR en duplicado en muestras de sangre seca de la primera semana de vida mejorará la E de los programas de SNFQ, disminuyendo el número de recién nacidos que precisan realizarse un test del sudor y/o genética y por tanto disminuyendo la ansiedad familiar asociada9. Existen estudios que muestran que la estrategia TIR/PAP podría ser viable en términos de costes por caso detectado, aunque las tasas de detección de formas clásicas de FQ podría ser menor en comparación con las estrategias TIR/ADN17. Según Seror et al. la tasa de detección de formas clásicas de FQ obtenidas con la estrategia TIR/PAP fue similar a la obtenida con TIR/ADN e TIR/PAP/ADN, pero la estrategia TIR/PAP requirió menos recursos y condujo a la detección de un número menor de formas leves en comparación con TIR/ADN17,18. El cambio de TIR/ADN a una estrategia TIR/PAP significaría renunciar a la posibilidad de identificar portadores de FQ17. La tabla 3 compara los resultados de diferentes programas de SNFQ.
Comparación de los resultados de los diferente protocolos de SNFQ
| Nuestro protocolo | TIR/TIR | TIR/p.508del | TIR/4 mutaciones | TIR/12 mutaciones | TIR/PAP (corte≥1ng/ml) | TIR/PAP (corte sugerido>0,9ng/ml) | TIR/ADN/TIR | TIR/PAP/ADN | |
|---|---|---|---|---|---|---|---|---|---|
| Sensibilidad | 85,71% | 86,6% | 89,9% | 71,4% | 9 95,8% | 85,7% | 92,8% | 90,48% | 71,4% |
| Especificidad | 99,78% | 99,4% | 99,9% | 99,9% | 99,9% | 99,9% | 99,9% | 99,91% | 99,9% |
| Valor predictivo positivo | 5,7% | 3,5% | 20,1% | 17,9% | 18,3% | 12,2% | 11,6 | 16,10 | 62,5% |
| Valor predictivo negativo | 99,9% | 99,9% | 99,9% | 99,9% | 99,9% | 99,9% | 99,9% |
El diagnóstico precoz del paciente con FQ mediante el SNFQ tiene muchos beneficios2,3,19. Cuando la FQ se diagnostica y trata precozmente, mejora el estado nutricional, la curva de crecimiento, el desarrollo cognitivo y la función pulmonar del lactante y disminuyen las colonizaciones crónicas, las exacerbaciones, la necesidad de ciclos de antibióticos y las hospitalizaciones2–4,19. La literatura indica que los pacientes con FQ diagnosticados dentro de los 2 primeros meses de vida es más probable que se beneficien de las intervenciones tempranas4,20. Nuestro programa de SNFQ ha permitido diagnosticar antes de las 8 semanas de vida al 79,45% de los pacientes diagnosticados de FQ desde la implantación del SNFQ, con una edad media al diagnostico de tan solo 39,86 días frente a los 258 días de vida de media de los pacientes no diagnosticados mediante el SNFQ. Este porcentaje es ligeramente superior al encontrado por otro autores como Kharrazi et al. que diagnostican al 74,5% de los recién nacidos con FQ antes de los 2 meses4.
Entre los hallazgos de los falsos negativos destaca la presencia de niveles bajos de TIR en 2 pacientes (16,67%) con IM, tal como reconocen otros autores1,2,21, pese a que en ambos casos los pacientes portaban mutaciones severas, concretamente uno F508del en homocigosis y otro en heterocigosis con p.lle507. En nuestra serie de casos hemos diagnosticado por SNFQ a 4 pacientes con IM, 3 de los cuales portaban en homocigosis la mutación F508del. Los neonatos con IM corren un mayor riesgo de ser falsos negativos del SNFQ que los neonatos sin IM, ya que en algunos casos los niveles de TIR en pacientes con IM son más bajos que en pacientes con FQ sin IM2. Por ello defendemos la importancia de que en el programa de SNFQ, tal y como tienen algunos países2, se informe de la presencia de IM para realizar a estos pacientes un test del sudor si la situación clínica del paciente lo permite y/o un estudio genético de FQ.
Entre las mutaciones encontradas en nuestra población destaca la presencia de F508del (c.1521_1523delCTT) en el 75% de los pacientes, porcentaje superior al encontrado por otros autores que consideran que dicha mutación representa aproximadamente el 60-70% de las mutaciones causantes de enfermedad en las poblaciones caucásicas de origen europeo1–3. En otros países como Reino Unido las más frecuentemente aisladas son F508del (74,1%), G551D [(c.1652G>A) (3,37%)] y G542X (1,85%)3; en Dinamarca la F508del asciende al 88%, frente al 50% de Italia y el 20% de Turquía. En las Islas Baleares es del 58,5% similar a Galicia y en el País Vasco asciende al 87%. La segunda mutación más frecuente ha sido la G542X, con una incidencia del 10,95%, similar a la encontrada en las Islas Baleares y significativamente superior a la de Asturias (5%), Castilla-León (3,8%) o Galicia (4,9%)3. L206W, (c.617T>G) ha sido aislada en el 4,1%, niveles más altos a los encontrados globalmente en España (1,6%), siendo más prevalente en la población hispánica (6,2%) y menos en afroamericanos (1,2%)3.
Mediante este estudio hemos podido conocer que la incidencia de la FQ en Andalucía es de 1:6.506, dato desconocido hasta el momento. La incidencia global en España, según datos de la Asociación Española del Cribado Neonatal (AECNE) es de 1:4.8073, aunque con variaciones entre las diferentes comunidades3. Así en Galicia y Catilla y León es de 1:4.439, 1:4.800 en Aragón, 1:6.244 en Cataluña y 1:6.602 en las Islas Baleares3,22. Prevemos que con la implantación del programa de SNFQ y el consiguiente consejo genético disminuirá la incidencia de la enfermedad en nuestra comunidad, tal y como ha ocurrido en otras regiones23.
Una de las principales fortalezas del presente estudio es el alto cumplimiento en el programa de SNFQ que ofrece una representación de la población de Andalucía. A pesar de que tenemos un tiempo de observación relativamente corto, los presentes resultados nos permiten reflexionar sobre posibles áreas de mejoras adicionales del algoritmo del SNFQ, que debe pasar por la introducción de estudio genético en nuestro programa SNFQ para así aumentar la S, disminuir la tasa de falsos positivos y disminuir la ansiedad familiar.
Conflicto de interesesLas autoras declaran no tener ningún conflicto de intereses.







