








Lung cancer (LC) remains a leading cause of cancer mortality worldwide, underscoring the urgent need for novel therapeutic targets. The integration of Mendelian randomization (MR) with proteomic data presents a novel approach to identifying potential targets for LC treatment.
MethodsThis study utilized a proteome-wide MR analysis, leveraging publicly available data from genome-wide association studies (GWAS) and protein quantitative trait loci (pQTL) studies. We analyzed genetic association data for LC from the TRICL-ILCCO Consortium and proteomic data from the Decode cohort. The MR framework was employed to estimate the causal effects of specific proteins on LC risk, supplemented by external validation, co-localization analyses, and exploration of protein–protein interaction (PPI) networks.
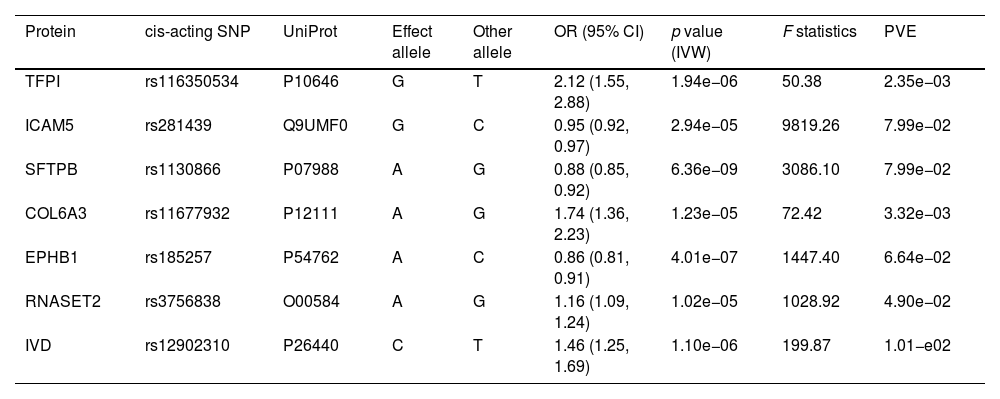
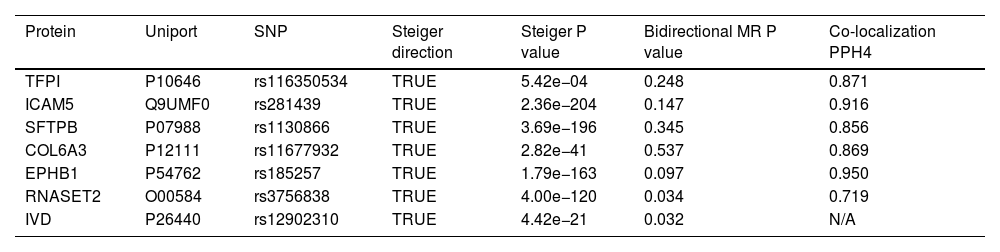
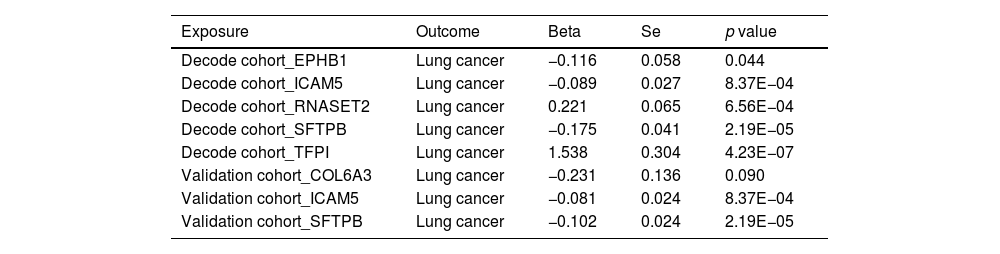
ResultsOur analysis identified five proteins (TFPI, ICAM5, SFTPB, COL6A3, EPHB1) with significant associations to LC risk. External validation confirmed the potential therapeutic relevance of ICAM5 and SFTPB. Co-localization analyses and PPI network exploration provided further insights into the biological pathways involved and their potential mechanistic roles in LC pathogenesis.
ConclusionThe study highlights the power of integrating genomic and proteomic data through MR analysis to uncover novel therapeutic targets for lung cancer. The identified proteins, particularly ICAM5 and SFTPB, offer promising directions for future research and development of targeted therapies, demonstrating the potential to advance personalized medicine in lung cancer treatment.








