










Lung cancer (LC) remains a leading cause of cancer mortality worldwide, underscoring the urgent need for novel therapeutic targets. The integration of Mendelian randomization (MR) with proteomic data presents a novel approach to identifying potential targets for LC treatment.
MethodsThis study utilized a proteome-wide MR analysis, leveraging publicly available data from genome-wide association studies (GWAS) and protein quantitative trait loci (pQTL) studies. We analyzed genetic association data for LC from the TRICL-ILCCO Consortium and proteomic data from the Decode cohort. The MR framework was employed to estimate the causal effects of specific proteins on LC risk, supplemented by external validation, co-localization analyses, and exploration of protein–protein interaction (PPI) networks.
ResultsOur analysis identified five proteins (TFPI, ICAM5, SFTPB, COL6A3, EPHB1) with significant associations to LC risk. External validation confirmed the potential therapeutic relevance of ICAM5 and SFTPB. Co-localization analyses and PPI network exploration provided further insights into the biological pathways involved and their potential mechanistic roles in LC pathogenesis.
ConclusionThe study highlights the power of integrating genomic and proteomic data through MR analysis to uncover novel therapeutic targets for lung cancer. The identified proteins, particularly ICAM5 and SFTPB, offer promising directions for future research and development of targeted therapies, demonstrating the potential to advance personalized medicine in lung cancer treatment.

Lung cancer (LC) has emerged as the leading cause of cancer-related mortality globally, with approximately 75% of patients diagnosed at an advanced stage.1 Although current therapies offer some benefits, the overall efficacy, particularly of chemotherapy, remains suboptimal, with response rates under 50%.2 Moreover, resistance to targeted therapies, exemplified by alterations in EGFR, RAS/RAF/PI3K, and mTOR pathways, represents a significant hurdle, undermining the effectiveness of existing treatments.3 Thus, there is an urgent need for innovative strategies that can overcome these challenges and improve patient outcomes.
The integration of genome-wide association studies (GWAS) with molecular biology offers a promising avenue for identifying and validating new therapeutic targets for LC. In this context, Mendelian randomization (MR) emerges as a powerful tool, using genetic variations as instrumental variables to infer causal relationships between potential drug targets and cancer outcomes.4–6 Such insights are invaluable for prioritizing targets with a stronger genetic rationale, potentially accelerating the transition from discovery to clinical application.7
Recent advances in proteomics and MR have opened new frontiers in oncology, enabling the identification of novel targets for a range of cancers, including prostate and breast malignancies.8,9 However, the application of these technologies in lung cancer, particularly through integrating protein quantitative trait loci (pQTL) data with GWAS findings, remains underexplored.
This study aims to bridge this gap by leveraging pQTL data from the Decode Consortium10 and patient data from the TRICL-ILCCO consortium11 to identify plasma proteins that could serve as viable therapeutic targets for LC. By employing a comprehensive analytical framework, including Bayesian co-localization, reverse causality assessment, and external validation with data from a European ancestry cohort12 and recent findings by Zheng et al.,13 this research endeavors to provide new insights into the molecular underpinnings of LC and identify potential avenues for therapeutic intervention.
MethodsOur investigation utilized a MR approach to identify potential therapeutic targets for LC, drawing upon publicly available proteomic and genomic data. Our analytical strategy is rooted in robust ethical standards, with all utilized data previously subjected to ethical approval and participant consent processes in their original studies.
Instrument construction and data acquisitionWe leveraged lung cancer genetic associations from 29,863 patients and 55,586 controls, provided by the TRICL-ILCCO Consortium.11 Proteomic data, encompassing 4907 plasma proteins from 35,559 participants, was sourced from the Decode cohort.10 Our criteria for pQTL selection were stringent, focusing on cis-pQTLs to ensure specificity and relevance to LC pathophysiology. Fig. 1 shows the framework of our research.
Mendelian randomization and validation processes
Employing the two-sample MR framework, we estimated the causal impact of identified proteins on LC risk, using Inverse Variance Weighted (IVW) methods for robust inference.14 Validation was pursued through external cohorts and additional MR analyses, with attention to the coherence and consistency of genetic instruments and their associations with LC risk.
Analytical rigor and secondary analysesTo assess the validity of our causal inferences, we conducted heterogeneity checks, Steiger filtering, and explored reverse causality scenarios.15 Concordance between protein functions and LC risk was further scrutinized via co-localization analysis, ensuring that observed associations were not artifacts of underlying genetic confounding.16,17
Functional insights and network analysesSingle-cell RNA sequencing data offered a nuanced view of protein expression in the lung microenvironment,18 while phenotype scanning provided context regarding the systemic relevance of these proteins.19 We integrated our results with protein–protein interaction (PPI) networks using databases like STRING and Drugbank to explore potential interactions and their therapeutic implications.20,21
For an in-depth description of our methodologies, including the criteria for pQTL selection, statistical analysis parameters, and detailed procedural steps, readers are referred to the Supplementary Material.
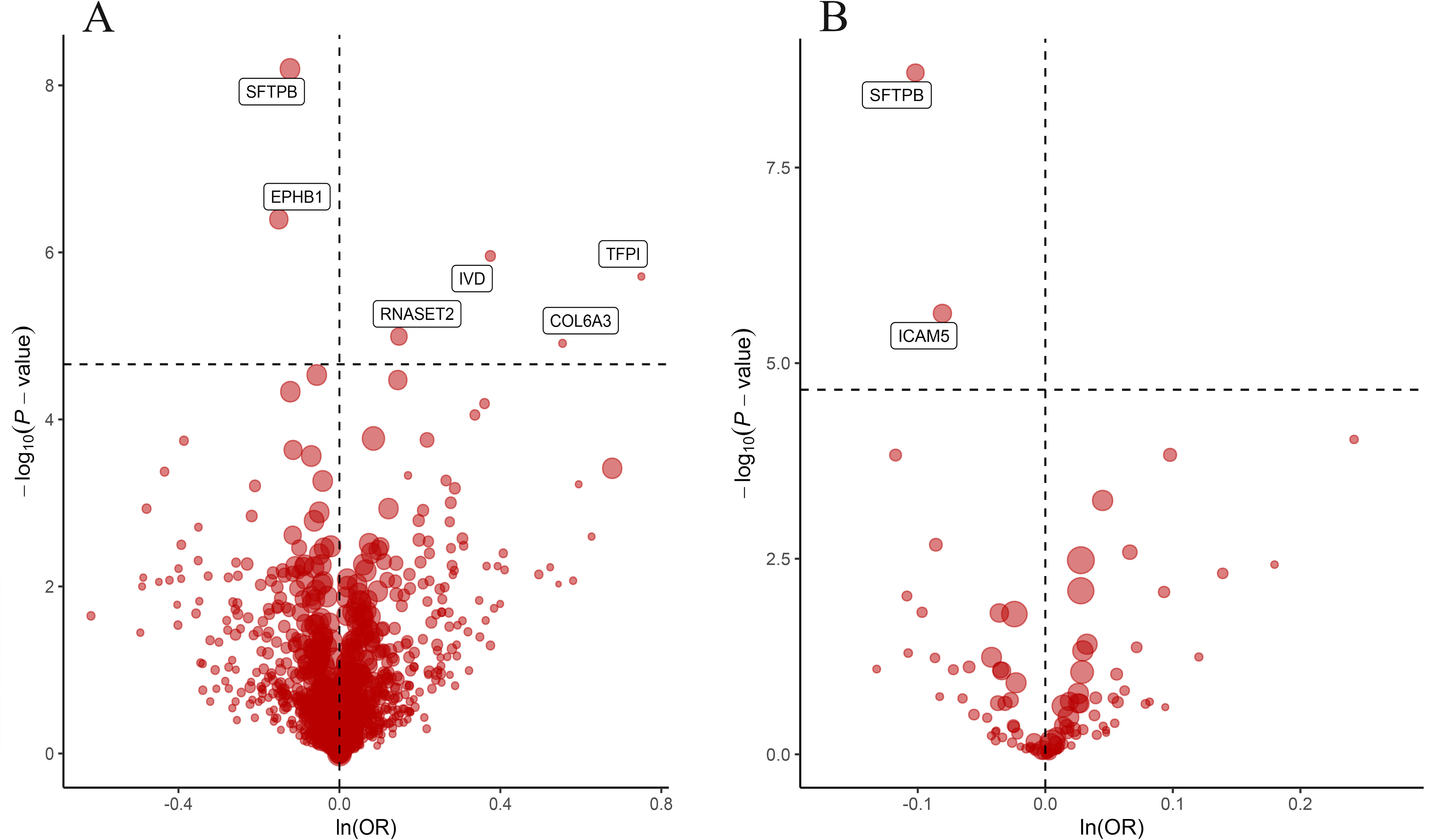
ResultsIdentification of lung cancer-associated proteins through proteome analysisOur rigorous application of the Bonferroni correction method unearthed significant associations between LC susceptibility and seven specific plasma proteins, as illustrated in our analytical outcomes (Table 1). These proteins include Tissue Factor Pathway Inhibitor (TFPI), Intercellular Adhesion Molecule 5 (ICAM5), Surfactant Protein B (SFTPB), Collagen Type VI Alpha 3 Chain (COL6A3), Ephrin Type-B Receptor 1 (EPHB1), Ribonuclease T2 (RNASET2), and Isovaleryl-CoA Dehydrogenase (IVD). Notably, elevated levels of ICAM5, SFTPB, and EPHB1 were associated with a reduced risk of LC, while higher concentrations of TFPI, COL6A3, RNASET2, and IVD correlated with increased LC risk. The consistency across analyses affirmed the absence of heterogeneity (Supplementary Table 1), bolstering the reliability of these protein-LC risk associations.
Mendelian randomization results for proteins of Decode cohort significantly related to lung cancer.
| Protein | cis-acting SNP | UniProt | Effect allele | Other allele | OR (95% CI) | p value (IVW) | F statistics | PVE |
|---|---|---|---|---|---|---|---|---|
| TFPI | rs116350534 | P10646 | G | T | 2.12 (1.55, 2.88) | 1.94e−06 | 50.38 | 2.35e−03 |
| ICAM5 | rs281439 | Q9UMF0 | G | C | 0.95 (0.92, 0.97) | 2.94e−05 | 9819.26 | 7.99e−02 |
| SFTPB | rs1130866 | P07988 | A | G | 0.88 (0.85, 0.92) | 6.36e−09 | 3086.10 | 7.99e−02 |
| COL6A3 | rs11677932 | P12111 | A | G | 1.74 (1.36, 2.23) | 1.23e−05 | 72.42 | 3.32e−03 |
| EPHB1 | rs185257 | P54762 | A | C | 0.86 (0.81, 0.91) | 4.01e−07 | 1447.40 | 6.64e−02 |
| RNASET2 | rs3756838 | O00584 | A | G | 1.16 (1.09, 1.24) | 1.02e−05 | 1028.92 | 4.90e−02 |
| IVD | rs12902310 | P26440 | C | T | 1.46 (1.25, 1.69) | 1.10e−06 | 199.87 | 1.01−e02 |
SNP: single-nucleotide polymorphism; OR: odds ratios; CI: confidence interval; PVE: proportion of variance explained; TFPI, Tissue Factor Pathway Inhibitor; ICAM5, Intercellular Adhesion Molecule 5; SFTPB, Surfactant Protein B; COL6A3, Collagen Type VI Alpha 3 Chain; EPHB1, Ephrin Type-B Receptor 1; RNASET2, Ribonuclease T2; IVD, Isovaleryl-CoA Dehydrogenase; IVW, inverse-variance weighted.
Subsequent sensitivity analyses, including Steiger filtering, reaffirmed the reliability of our MR findings, underscoring a consistent causal directionality (Table 2). Bidirectional MR analysis revealed on causal relationship between LC and the protein levels of TFPI, ICAM5, SFTPB, COL6A3, or EPHB1 (all P > 0.05). RANSET2 revealed undefined directional causal effects with a P value of 0.034, 0.026, and 0.781 in IVW, MR-Egger and weighted median model, respectively. IVD exhibited a reverse causal effect with a P value of 0.032, 0.041, and 0.005 in IVW, MR-Egger and weighted median method, respectively. To further refine causal credit, we excluded IVD proteins from subsequent analyses. Bayesian co-localization then confirmed the shared genetic variations linked to LC risk, offering a robust foundation for their causal inference (Supplementary Fig. 2).
Overview of Steiger filtering analyses, Bayesian co-localization analysis, and reverse causality detection on seven candidate target proteins.
| Protein | Uniport | SNP | Steiger direction | Steiger P value | Bidirectional MR P value | Co-localization PPH4 |
|---|---|---|---|---|---|---|
| TFPI | P10646 | rs116350534 | TRUE | 5.42e−04 | 0.248 | 0.871 |
| ICAM5 | Q9UMF0 | rs281439 | TRUE | 2.36e−204 | 0.147 | 0.916 |
| SFTPB | P07988 | rs1130866 | TRUE | 3.69e−196 | 0.345 | 0.856 |
| COL6A3 | P12111 | rs11677932 | TRUE | 2.82e−41 | 0.537 | 0.869 |
| EPHB1 | P54762 | rs185257 | TRUE | 1.79e−163 | 0.097 | 0.950 |
| RNASET2 | O00584 | rs3756838 | TRUE | 4.00e−120 | 0.034 | 0.719 |
| IVD | P26440 | rs12902310 | TRUE | 4.42e−21 | 0.032 | N/A |
SNP: single-nucleotide polymorphism; TFPI, Tissue Factor Pathway Inhibitor; ICAM5, Intercellular Adhesion Molecule 5; SFTPB, Surfactant Protein B; COL6A3, Collagen Type VI Alpha 3 Chain; EPHB1, Ephrin Type-B Receptor 1; RNASET2, Ribonuclease T2; IVD, Isovaleryl-CoA Dehydrogenase; N/A, not applicable.
By leveraging additional datasets for external validation, we corroborated the relevance of EPHB1, ICAM5, RNASET2, SFTPB, and TFPI to LC risk, echoing findings from an independent cohort study by Battram et al. The robust associations affirmed through significant single nucleotide polymorphisms (SNPs) in the validation cohorts (Table 3, Supplementary Fig. 3) particularly emphasized the roles of ICAM5 and SFTPB, underscoring their therapeutic potential (Fig. 2).
External validation of selected protein-lung cancer correlations using mendelian randomization analysis.
| Exposure | Outcome | Beta | Se | p value |
|---|---|---|---|---|
| Decode cohort_EPHB1 | Lung cancer | −0.116 | 0.058 | 0.044 |
| Decode cohort_ICAM5 | Lung cancer | −0.089 | 0.027 | 8.37E−04 |
| Decode cohort_RNASET2 | Lung cancer | 0.221 | 0.065 | 6.56E−04 |
| Decode cohort_SFTPB | Lung cancer | −0.175 | 0.041 | 2.19E−05 |
| Decode cohort_TFPI | Lung cancer | 1.538 | 0.304 | 4.23E−07 |
| Validation cohort_COL6A3 | Lung cancer | −0.231 | 0.136 | 0.090 |
| Validation cohort_ICAM5 | Lung cancer | −0.081 | 0.024 | 8.37E−04 |
| Validation cohort_SFTPB | Lung cancer | −0.102 | 0.024 | 2.19E−05 |
TFPI, Tissue Factor Pathway Inhibitor; ICAM5, Intercellular Adhesion Molecule 5; SFTPB, Surfactant Protein B; COL6A3, Collagen Type VI Alpha 3 Chain; EPHB1, Ephrin Type-B Receptor 1; RNASET2, Ribonuclease T2.
Our deep dive into single-cell RNA sequencing data revealed nuanced expressions of ICAM5 and SFTPB within diverse lung cell populations, offering a granular view of their biological milieu. Despite ICAM5s ubiquitous presence, SFTPB's enrichment in alveolar type II cells highlighted its specific pathophysiological context within lung tissue, spotlighting its potential as a therapeutic target (Fig. 3).
Integrative analysis with pharmaceutical interventions
Our investigation extended to delineate the connectivity between our identified proteins and established LC therapeutic pathways. Particularly, interactions between TFPI and VCAM1, as well as EPHB1's association with the Eph/Ephrin signaling axis, unveiled potential novel intervention points. While EPHB1 has already garnered attention for its therapeutic applicability, COL6A3's interaction with known LC targets signals uncharted therapeutic territory, warranting further exploration (Fig. 4, Supplementary Table 3).
Discussion
Our pioneering study leverages blood proteome data alongside bidirectional Mendelian randomization and Bayesian co-localization to delineate potential therapeutic proteins implicated in lung cancer pharmacodynamics. Among the identified candidates—TFPI, ICAM5, SFTPB, COL6A3, and EPHB1—ICAM5 and COL6A3 have been corroborated in external cohorts, underlining their therapeutic relevance. ICAM5 emerges as a novel target, heretofore unexplored in the context of LC therapy, thereby opening new investigational avenues.
The integration of genetic insights to ascertain drug target efficacy signifies a paradigm shift in pharmacological innovation, as genetically validated targets demonstrate enhanced success rates in drug development.4,7 Through meticulous MR and co-localization analyses, our investigation validates several proteins associated with LC pathogenesis, substantiating their roles as prospective therapeutic targets grounded on robust genetic evidence.10,11,13
Despite rigorous MR scrutiny across large patient cohorts, our analysis acknowledges inherent methodological constraints, such as the risk of horizontal pleiotropy or confounders influencing genetic instrumental variables. Nonetheless, the careful exclusion of reverse causality, especially highlighted by the distinct roles of TFPI, ICAM5, SFTPB, COL6A3, and EPHB1, reinforces their relevance in LC etiology.
Interestingly, TFPI, associated with thrombosis and inflammation, holds promise beyond its conventional biological roles, suggesting potential anti-tumor activity that warrants further exploration in LC contexts.22–26 Concurrently, the associations between LC risk and other proteins like RNASET2 underscore intricate interplays between inflammatory pathways and cancer progression, suggesting multifaceted roles in tumor biology.
The therapeutic landscape of LC, particularly immunotherapy, remains fraught with challenges, notably the limited efficacy in certain patient subsets.27 Our findings illuminate potential interactions between identified proteins and known LC targets, suggesting alternative therapeutic strategies. For instance, EPHB1's linkage to the Eph/Ephrin signaling implicates it in key cancer processes, advocating its potential as an actionable target.28–32
Additionally, our exploratory analyses intimate at the utility of SFTPB and ICAM5 as putative markers and modulators within the LC microenvironment, potentially informing targeted therapeutic interventions.33,34 Such insights not only advance our understanding of LC biology but also chart promising directions for future drug development, emphasizing precision medicine's pivotal role in oncology.
Our study has several limitations. Firstly, the GWAS data utilized in our analysis were obtained from diverse large-scale sequencing studies, and variations in the study protocols across different cohorts might introduce bias. Secondly, our research primarily focused on the European populations, making it challenging to generalize our findings to other ethnic ancestry. Nevertheless, we conducted an extensive population-based validation study including the UK and Finnish populations. More studies in non-European ancestry needed to be further explored to translate these promising drug targets into clinical application.
ConclusionOur study elucidates the significant associations between LC risk and the levels of specific proteins, notably TFPI, ICAM5, SFTPB, COL6A3, EPHB1, and RNASET2, through proteome-wide Mendelian randomization analysis. These findings not only spotlight novel therapeutic targets, particularly ICAM5 and SFTPB, but also underscore the necessity for further mechanistic studies to fully understand their roles in LC pathogenesis and treatment. By providing a genetic underpinning for these potential targets, our research paves the way for their future application in developing more precise and effective LC therapies, heralding a new era of genetically informed drug discovery in oncology.
Ethics approval and consent to participateEthical approval and informed consent and were not required for this study, as we utilized publicly accessible summary data, and ethics approval and participant consent had already been obtained in the original GWAS.
Funding sourcesThis work was supported by The Beijing Natural Science Foundation (7232134); National Key R&D Program of China (2022YFC2407404); Special Research Fund for Central Universities, Peking Union Medical College (2022-I2M-C&T-B-065, 2022-I2M-C&T-B-060); National High-Level Hospital Clinical Research Funding (2022-PUMCH-A-018, 2022-PUMCH-C-043); Beijing Municipal Science & Technology Commission (Z211100002921058).
Authors’ contributionsKun Wang and Hang Yi conceived and designed the study. Kun Wang, Hang Yi, Yan Wang and Donghui Jin contributed to the writing of the manuscript. Kun Wang performed formal analysis and visualization. Yousheng Mao was responsible for investigations. Kun Wang, Hang Yi, Yan Wang, Donghui Jin, Guochao Zhang, and Yousheng Mao participated in the analysis and discussion of the data. All the authors revised the article critically and approved the final version. Kun Wang and Hang Yi contributed equally to this work as co-first authors.
Consent for publicationNo conflict of interest exits in the submission of this manuscript, and manuscript is approved by all authors for publication.
Conflict of interestsNo disclosures to report.
Availability of data and materialsdeCODE Genetics whole-genome sequencing variants was available in the European Variation Archive (registration ID: PRJEB15197; Access Link: https://download.decode.is/form/folder/proteomics; Note: Access to the raw data requires registration using an academic email address and a formal application for access). WGAS data of TRICL-ILCCO Consortium was available in https://gwas.mrcieu.ac.uk/files/ieu-a-987/ieu-a-987.vcf.gz (Dataset ID: ieu-a-987). TwoSampleMR R package (v0.5.6; https://github.com/mrcieu/TwoSampleMR) could perform Two Sample MR analysis. Co-localization analysis was carried out using GALAXY of BioInfoTools based on the coloc package (https://biowinford.site:3838/OnlineTools1/).














