

Chronic obstructive pulmonary disease (COPD) is characterized by restricted airflow. The best-documented genetic factor is alpha-1 antitrypsin (AAT). AAT is encoded by the SERPINA1 gene. The PiZ (rs28929474) and PiS (rs17580) variants are believed to cause severe AAT deficiency and are linked to a high risk of developing COPD. This study sought to identify whether genetic polymorphisms rs28929474 and rs17580 are associated with COPD susceptibility and lung function values in a Mexican mestizo population.
MethodsIn this study, 558 smokers were included, of whom 279 had COPD and 279 did not (smokers without COPD – SWC). The PiS and PiZ variants were genotyped by allelic discrimination. Independent populations and lung function values were compared using the Kruskal–Wallis test. A bivariate logistic regression analysis was also conducted.
ResultsStage I and IV COPD patients showed significant differences in the frequencies of both heterozygous genotypes compared to SWC. For PiS, individuals with the heterozygous genotype AT demonstrated a decreased FEV1/FVC ratio compared to subjects with the homozygous genotype AA (P=.037). A significant association was found between the FEV1/FVC ratio and genotype AA for PiS (OR=0.982, β coefficient=−0.019, 95% CI=0.966–0.997).
ConclusionsCOPD-causing AAT deficiency risk alleles exist at a very low frequency among Mexican mestizo population. Although they are not directly linked in our study population with disease susceptibility, these risk alleles are associated with poorer lung function measurements. It is important to characterize how often these genetic risk variants occur in other Latin American populations.
La enfermedad pulmonar obstructiva crónica (EPOC) se caracteriza por dificultad para respirar. El factor genético mejor documentado es la deficiencia de alfa-1 antitripsina (A1AT). La A1AT está codificada por el gen SERPINA1. Se considera que las variantes PiZ (rs28929474) y PiS (rs17580) causan una deficiencia grave de A1AT y que están relacionadas con un alto riesgo de desarrollar EPOC. En este estudio se busca identificar si los polimorfismos genéticos rs28929474 y rs17580 conllevan a la predisposición a la EPOC y su relación con los valores de función pulmonar en la población mestiza mexicana.
MétodosPara el estudio actual se incluyeron 558 fumadores, de los cuales 279 padecían EPOC y 279 no (fumadores sin EPOC [FSE]). Se genotiparon las variantes PiS y PiZ por discriminación alélica. Se evaluó la comparación entre poblaciones independientes y los valores de función pulmonar mediante la prueba de Kruskal–Wallis. Además, se realizó un análisis de regresión logística bivariada.
ResultadosLos pacientes con EPOC en estadio i y iv presentaron diferencias significativas en cuanto a las frecuencias de ambos genotipos heterocigotocigotos en comparación con los FSE. Para PiS, los sujetos con el genotipo heterocigotocigoto AT presentaron una reducción del cociente FEV1/FVC en comparación con los sujetos con el genotipo homocigoto AA (p=0,037). Se detectó una relación significativa entre el valor FEV1/FVC y el genotipo AA para PiS (OR=0,982; coeficiente=–0,019; IC 95%=0,966–0,997).
ConclusionesLos alelos con riesgo de deficiencia de A1AT que causan EPOC son poco frecuentes entre la población mestiza mexicana. Aunque en nuestra población de estudio no tienen relación directa con la predisposición genética a la enfermedad, estos alelos de riesgo se asocian a peores niveles de función pulmonar. Es importante describir con qué frecuencia aparecen estas variantes genéticas de riesgo en otras poblaciones latinoamericanas.
The Global Initiative for Chronic Obstructive Lung Disease (GOLD) defines chronic obstructive pulmonary disease (COPD) as a preventable and treatable condition with extrapulmonary effects that contribute to severity. The pulmonary disease component is characterized by restricted airflow, which is not completely reversible. Airflow obstruction is usually progressive and is associated with an abnormal inflammatory response in the lung, caused by particles or toxic gases. COPD is a disease with several genetic components, of which alpha-1 antitrypsin (AAT) deficiency is the best documented to date.1
AAT is a protein that is mainly synthesized in the liver and secreted into the blood stream, although it is also synthesized to a lesser degree by alveolar macrophages. AAT is an important protease inhibitor, with normal plasma levels ranging from 120 to 220mg/dL, and is also the enzyme responsible for maintaining protease-antiprotease equilibrium in the lung.2 Recent studies have indicated that AAT has anti-inflammatory activity,3 inhibits TNF gene expression,4 and inhibits human monocyte and neutrophil migration when activated by lipopolysaccharide in vitro.5
AAT is encoded by the SERPINA1 gene, which is located in the q32.1 cytogenetic band of chromosome 14. It is transmitted in an autosomal codominant fashion and is characterized by large numbers of polymorphic variants.6,7 The group of AAT variants is referred to as the protease inhibitor (Pi) system. Most of these variants lack any clinical relevance, and only 30 have a defined pathological impact.8 The most common variant is PiM, which is considered a “normal phenotype”, given that 90% of healthy subjects have the homozygous genotype (PiMM). The PiZ (rs28929474) and PiS (rs17580) variants give rise to 90% of AAT deficiency cases. When combined, these variants produce the PiSS, PiSZ and PiZZ phenotypes, the latter two of which are considered severely deficient variants and are associated with low protein concentrations in serum and a high risk of developing COPD at an early age.9,10 Variant Z arises from a G-to-A transition at nucleotide 11940 in exon 5 of the SERPINA1 gene, which results in the substitution of glutamic acid with lysine at position 366 of the AAT and gives rise to a protein with abnormal antiproteolytic function.10,11 Variant S arises from an A-to-T transversion at nucleotide 9628 in exon 3, which leads to the substitution of glutamic acid with valine at position 288 of the protein.11–13
Genotyping the clinically relevant SERPINA1 variants will be beneficial not only for identifying subjects with severe AAT deficiency but also for identifying cases with intermediate deficiency. The latter type of deficiency has also been linked to a decline in lung function.14
In this study, we analyzed the frequency of the genetic polymorphisms PiZ (rs28929474) and PiS (rs17580) associated with AAT deficiency and their association with lung function values in a Mexican mestizo population.
MethodsParticipantsAn exploratory prospective study was conducted from November 2008 to August 2012; 279 smokers without COPD (SWC) were recruited from a smoking cessation support clinic, and 279 patients with COPD were recruited from the COPD clinic. Both clinics belong to the Instituto Nacional de Enfermedades Respiratorias Ismael Cosío Villegas (INER) of Mexico. All subjects were over 40 years of age, Mexican mestizos by ancestry (parents and grandparents born in Mexico), smokers or ex-smokers with a minimum 10 pack-year history of smoking for 10 or more years, and diagnosed by pulmonology specialists at the INER COPD clinic. Diagnosis was based on medical history, physical examination, and spirometry data, taking into account the criteria established by the American Thoracic Society (ATS). The spirometer used was model Vmax 2130 (Sensormedics, Yorba Linda, CA, USA), and it was calibrated with a 3-L syringe at a pressure of 588mmHg (the altitude of Mexico City). The values predicted by Pérez Padilla15 were used, which were ideal for the Mexican population. For the post-bronchodilator test, 400mg of salbutamol was administered using an aerosol inhaler and spacer. Spirometry technicians were certified by the National Institute for Occupational Safety and Health (NIOSH). Smokers without COPD (SWC) served as controls. Following the GOLD diagnostic criteria, the control subjects had a post-bronchodilator FEV1/FVC ratio equal to or greater than 70%, while the cases diagnosed with COPD had a FEV1/FVC ratio of less than 70%. Subjects diagnosed with bronchial asthma, bronchiectasis, active tuberculosis, lung cancer, cystic fibrosis, hypersensitivity pneumonitis, or idiopathic pulmonary fibrosis were excluded. A 6-mL sample of peripheral blood was collected in tubes containing EDTA as anticoagulant. All participants filled out a questionnaire on anthropometric data and any history of inherited pathologies. Subjects voluntarily agreed to participate and signed an informed consent letter created specifically for this study. The protocol was approved by INER science and research bioethics and biosecurity committees.
Genomic DNA Extraction and Concentration AdjustmentPeripheral blood cells were obtained by venous puncture. Genomic DNA was extracted using the commercially available BDtract DNA isolation kit (Maxim Biotech, San Francisco CA, USA). DNA was quantified by UV light microspectrophotometry at 260nm using an ACTGene spectrophotometer (ACTGene, Inc., New Jersey, USA). Protein contamination was determined at 280nm, and a sample was considered free of contaminants when the 260/280 ratio was between 1.7 and 2.0. Each sample concentration was adjusted to 25ng/μL for subsequent genotyping.
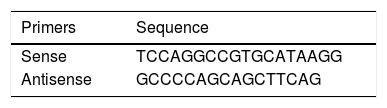
PiS and PiZ Allele GenotypingSamples were genotyped by allelic discrimination using commercially available TaqMan probes and a 7300 Real Timer PCR System thermocycler (Applied Biosystems, CA, USA). For rs17580 (PiS variant), a probe pre-designed by the manufacturer was used (ID: C_594695_20, Applied Biosystems, CA, USA). For rs28929474 (PiZ variant), probes and primers designed by Bartels et al.,11 were used, which are shown in Table 1. Conditions for genotyping both alleles were optimized in the laboratory. The cycling conditions for both probes were as follows: pre-read at 50°C for 1min; absolute quantification at 50°C for 2min, followed by 1 cycle at 95°C for 10min, 1 cycle at 95°C for 15s, and 40 cycles at 60°C for 1min; and post-read at 50°C for 1min. Genotypes were assigned taking allelic discrimination into consideration and were confirmed using absolute quantification. Additionally, 4 controls without a template (contamination controls) were included in each genotyping plate. The data were interpreted using Sequence Detection Software (SDS v. 1.4, Applied Biosystems, CA, USA). For both SNPs, fluorophores used were VIC for allele A and FAM for allele B.
Statistical AnalysisThe statistics program SPSS v. 15.0 for Windows was used to describe the study population and determine the median, minimum, and maximum values for each variable. Significant differences between genotypic and allelic frequency (AF) values were calculated with Epi Info version 6.04d.16 Haploview 4.2 was used to assess Hardy–Weinberg (HW) equilibrium for the polymorphisms and generate the haplotype.17 Comparisons between independent populations, based on the genotypes obtained and lung function values, were estimated using the Kruskal–Wallis test. In addition, a bivariate logistical regression analysis was conducted.
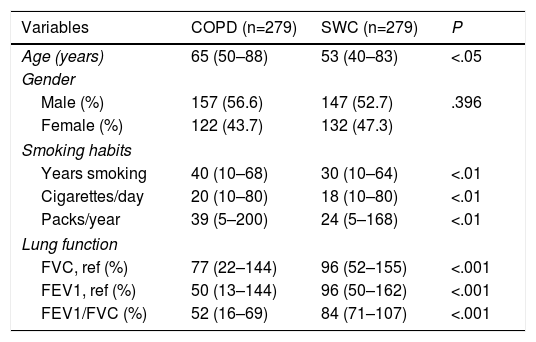
ResultsThe characteristics of both study groups are briefly summarized in Table 2. A statistically significant difference was found when the age of smokers with COPD was compared to the age of smokers without COPD (P<.05). Gender representation was evenly distributed between groups. When smoking habits between patients with COPD and SWCs were compared, the patients were found to smoke more cigarettes and have decreased lung function in comparison to the control group.
Age, Gender, Smoking Habits, and Lung Function Data for COPD Patients and Healthy Smokers.
| Variables | COPD (n=279) | SWC (n=279) | P |
|---|---|---|---|
| Age (years) | 65 (50–88) | 53 (40–83) | <.05 |
| Gender | |||
| Male (%) | 157 (56.6) | 147 (52.7) | .396 |
| Female (%) | 122 (43.7) | 132 (47.3) | |
| Smoking habits | |||
| Years smoking | 40 (10–68) | 30 (10–64) | <.01 |
| Cigarettes/day | 20 (10–80) | 18 (10–80) | <.01 |
| Packs/year | 39 (5–200) | 24 (5–168) | <.01 |
| Lung function | |||
| FVC, ref (%) | 77 (22–144) | 96 (52–155) | <.001 |
| FEV1, ref (%) | 50 (13–144) | 96 (50–162) | <.001 |
| FEV1/FVC (%) | 52 (16–69) | 84 (71–107) | <.001 |
Median and minimum and maximum values are shown. SWC: smokers without COPD.
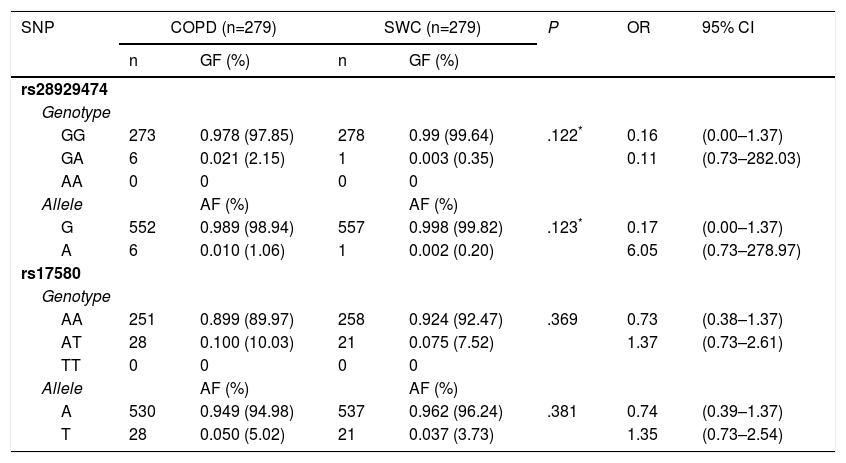
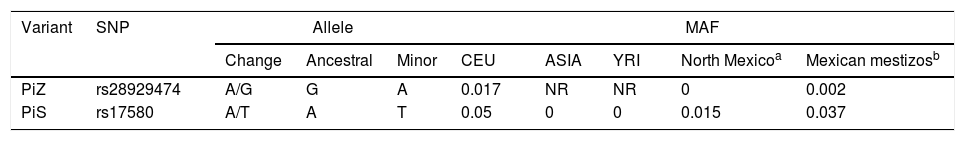
The genotypic and allelic frequencies (GF and AF, respectively) of polymorphisms rs17580 (PiS) and rs28929474 (PiZ) are shown in Table 3. No statistically significant difference for either genotype or allele was found when both groups were compared. The genetic data for the studied polymorphisms, as well as a comparison between the allelic frequencies for both SNPs, are shown in Table 4. AF comparison was performed between a population from northern Mexico,18 three populations reported in HapMap,19 and our SWC group representing Mexican mestizos. For rs17580, the minor allele was found at an AF of 0.05 (5%) in the Caucasian population but was not detected in the Asian or Yoruban populations. In contrast, when the Mexican population studied by Sánchez-Domínguez18 was compared to our control group, different AFs for the T allele (0.015 vs 0.037) were found. Out of all of the populations studied in HapMap, the rs28929474 was present only in the Caucasian population (AF=0.017, 1.7%). As previously reported, the minor allele was not found in the population from northern Mexico, while in our control group, this allele only reached an AF of 0.002 (0.02%).
Genotypic and Allelic Frequencies for rs28929474 (PiZ) and rs17580 (PiS) in COPD Patients and Control Smokers Without COPD.
| SNP | COPD (n=279) | SWC (n=279) | P | OR | 95% CI | ||
|---|---|---|---|---|---|---|---|
| n | GF (%) | n | GF (%) | ||||
| rs28929474 | |||||||
| Genotype | |||||||
| GG | 273 | 0.978 (97.85) | 278 | 0.99 (99.64) | .122* | 0.16 | (0.00–1.37) |
| GA | 6 | 0.021 (2.15) | 1 | 0.003 (0.35) | 0.11 | (0.73–282.03) | |
| AA | 0 | 0 | 0 | 0 | |||
| Allele | AF (%) | AF (%) | |||||
| G | 552 | 0.989 (98.94) | 557 | 0.998 (99.82) | .123* | 0.17 | (0.00–1.37) |
| A | 6 | 0.010 (1.06) | 1 | 0.002 (0.20) | 6.05 | (0.73–278.97) | |
| rs17580 | |||||||
| Genotype | |||||||
| AA | 251 | 0.899 (89.97) | 258 | 0.924 (92.47) | .369 | 0.73 | (0.38–1.37) |
| AT | 28 | 0.100 (10.03) | 21 | 0.075 (7.52) | 1.37 | (0.73–2.61) | |
| TT | 0 | 0 | 0 | 0 | |||
| Allele | AF (%) | AF (%) | |||||
| A | 530 | 0.949 (94.98) | 537 | 0.962 (96.24) | .381 | 0.74 | (0.39–1.37) |
| T | 28 | 0.050 (5.02) | 21 | 0.037 (3.73) | 1.35 | (0.73–2.54) | |
Genotypic and allelic frequencies are shown as absolute and percentage data. GG and AA genotypes correspond to PiM variant, GA and AA are genotypes coding to PiZ, while AT and TT code to the PiS variant. CI: 95% confidence interval; GF: genomic frequency; OR: odds ratio; SWC: smokers without COPD.
Comparison of Minor Allele Frequency Between Populations Reported in HapMap, a Population From Northern Mexico, and our Control Group.
| Variant | SNP | Allele | MAF | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Change | Ancestral | Minor | CEU | ASIA | YRI | North Mexicoa | Mexican mestizosb | ||
| PiZ | rs28929474 | A/G | G | A | 0.017 | NR | NR | 0 | 0.002 |
| PiS | rs17580 | A/T | A | T | 0.05 | 0 | 0 | 0.015 | 0.037 |
MAF: minor allele frequency; CEU: Caucasian; YRI: Nigerian Yoruba; NR: not reported.
HW equilibrium and haplotype formation were determined using Haploview 4.2, and both rs17580 and rs28949474 were found to be in HW equilibrium (P=.7277 and 1.0, respectively). In terms of haplotypes, we did not find any common haplotype structure between the case and control populations evaluated in this study (r2<0.80).
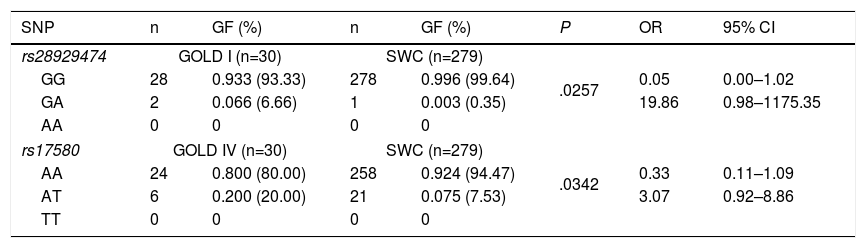
Statistical analysis of genetic association showed statistically significant differences between GOLD stages I and IV in patients with COPD for both heterozygous genotypes. However, the confidence intervals were broad and included the null value (1.0), specifically for rs28929474 (PiZ), and the heterozygote frequency was too low in both study groups (Table 5). No statistically significant differences were found when stages II and III were compared (data not shown).
Genotypic Frequencies in COPD Patients With GOLD Stage I or IV Disease and Control Smokers Without COPD.
| SNP | n | GF (%) | n | GF (%) | P | OR | 95% CI |
|---|---|---|---|---|---|---|---|
| rs28929474 | GOLD I (n=30) | SWC (n=279) | |||||
| GG | 28 | 0.933 (93.33) | 278 | 0.996 (99.64) | .0257 | 0.05 | 0.00–1.02 |
| GA | 2 | 0.066 (6.66) | 1 | 0.003 (0.35) | 19.86 | 0.98–1175.35 | |
| AA | 0 | 0 | 0 | 0 | |||
| rs17580 | GOLD IV (n=30) | SWC (n=279) | |||||
| AA | 24 | 0.800 (80.00) | 258 | 0.924 (94.47) | .0342 | 0.33 | 0.11–1.09 |
| AT | 6 | 0.200 (20.00) | 21 | 0.075 (7.53) | 3.07 | 0.92–8.86 | |
| TT | 0 | 0 | 0 | 0 | |||
SNP nomenclature: rs28929474 corresponds to serum PiZ variant, while rs17580 to PiS. GG and AA genotypes correspond to PiM variant, GA and AA are genotypes coding to PiZ, while AT and TT code to PiS variant. CI: confidence interval; GF: genotypic frequency; OR: odds ratio; SWC: smokers without COPD.
The study population (COPD and SWC) was divided according to the genotypes detected, yielding subjects with the common genotype and subjects with the risk genotype for each SNP. The Kruskal–Wallis test was used to compare lung function values. When median FEV1/FVC was compared, the heterozygous genotype for rs17580 (PiS) exhibited a P value of .037 (Fig. 1). The box and whisker plot, which indicates the median for each group and its respective limits, shows that individuals with the heterozygous genotype AT had a lower FEV1/FVC ratio compared to subjects with the homozygous genotype AA, which corresponds to PiM variant in the serum. Furthermore, a bivariate logistical regression revealed a significant association between the FEV1/FVC ratio and the AA genotype for rs17580 (OR=0.982, β coefficient=−0.019, 95% CI=0.966–0.997). These results suggest that genotypes other than AA are risk factors, since the AA genotype encodes normal protein. We found no association with any other lung function measurements.
Discussion based on the genotypes obtained for rs17580 (PiS) in the study population. Note: Generated with SPSS 15.0 for Windows using the Kruskal–Wallis test.")
COPD development in an AAT deficiency context is generally variable. Host-specific factors, such as gene modifications, probably interact with environmental factors to contribute to individual disease manifestations. Our study was the first to evaluate the frequency of 2 main risk alleles for AAT (SERPINA1) deficiency, PiS (rs17580) and PiZ (rs28929474), in a Latin American mestizo population with Amerindian and Caucasian genetic contributions. We studied 558 smokers, who were divided according to health status into patients with COPD and disease-free smokers. Our findings indicated that the frequency of the risk genotypes (TT and AA homozygotes) was zero, while the PiS heterozygote (rs17580 AT) genotype was detected at a frequency of up to 10% in patients with COPD and 7.52% in disease-free smokers. In contrast, the PiZ heterozygous genotype (rs28929474 GA) was found in 2.15% of patients but only in 0.35% of control group subjects. The differences in risk genotype frequencies are almost certainly due to the ancestral origin of each genetic variable.
Smoking is the main cause of more than 90% of COPD cases. However, it has been estimated that only 10%–20% of smokers develop this disease, and this could be due to genetic and/or environmental factors that modulate the toxic effect of cigarette smoke.20 The PLATINO study showed that COPD prevalence in Latin America ranges from 5.6% in Mexican women to 27.2% in Uruguayan men, with the lowest prevalence of the disease reported in Mexico. One hypothesis attributes this difference to the altitude above sea level.21,22 However, the population of Mexico City differs from that of other cities mainly due to its ethnic composition, which suggests that genetic factors could play a role in the low COPD prevalence observed among Mexicans. In this regard, AAT deficiency is the genetic factor most clearly associated with COPD and increases the risk of developing pulmonary emphysema for smokers, especially at an early age.20 The guidelines of the World Health Organization (WHO) and the ATS/ERS scientific societies have explicitly indicated that quantification of serum AAT levels should be performed for all COPD patients as part of the standard diagnostic procedure. It is also important to indicate when and how other laboratory diagnostic tests, such as phenotype or genotype assessments, should be performed.23 However, serum AAT quantification requires the absence of temporary inflammatory processes, which could alter the AAT levels.
It is estimated that 100000 individuals are affected by AAT deficiency in the United States, and similar rates have been reported in Europe.24 There are at least 116 million carriers (PiMS and PiMZ) and 3.4 million individuals with a combination of deficiency alleles (PiSS, PiSZ, PiZZ). In addition, it has been reported that AAT deficiency affects individuals of all racial sub-groups worldwide.25 However, there are few data concerning the frequency of these deficiency alleles in a Mexican mestizo population. For historic reasons, this population contains a large, mainly Spanish, Caucasian genetic component.26 In this respect, the use of racial or ethnic classifications in medicine has been the subject of multiple studies.27 Currently, lung function measurements are one of the few clinical applications in which race definition (in terms of ethnicity) is taken into account to establish health condition reference parameters, and ancestry-based lung function prediction models have been shown to fit the data better than standard models.28
Genome mapping studies have suggested that the PiZ allele arose in northern Europe29,30 and it is estimated that this variant has been in existence for 107–135 generations. The PiS allele has been in existence for 279–470 generations, and it is thought to have originated in the European region, given its high incidence in the Iberian Peninsula.30,31
Poorer lung function measurements associated with PiZ homozygotes are well documented. However, the role of the so-called intermediate deficiency genotypes (PiMZ and PiMS) has not been well studied. In our study, we found a lower FEV1/FVC (%) value among AT heterozygote carriers for rs17580 (PiS) compared to AA homozygous subjects. Other studies have shown a similar effect for the PiZ genotype. First in 200131 and subsequently in 2002,32 Morten Dahl et al. studied the effect of the intermediate deficiency genotypes on lung function in a Danish population. By taking spirometric measurements (FEV1 and FVC) and genotyping SERPINA1 alleles, these authors found that PiSZ heterozygotes had lower FEV1/FVC ratios compared to individuals without the risk genotype (PiMM). Although PiZ homozygotes had lower FEV1 percentages and FEV1/FVC ratios than the rest of the genotypes (MS and MZ), stratification of the results by smoking status revealed that the decrease in lung function in SZ and ZZ vs MM individuals was only statistically significant between smokers and ex-smokers. Furthermore, MZ individuals were shown to have decreased lung function compared to MM patients, which suggests that the MZ heterozygous genotype (rs28929474 GA) only acts in certain contexts, which have yet to be determined. These studies show that the MZ genotype, compared to the MM genotype, is associated with decreased lung function in individuals with COPD. Furthermore, the SZ and ZZ genotypes are associated with airway obstruction and decreased lung function, especially in smokers.31
Proper examination of population subgroups is an vital step for identifying important factors related to the prevention and timely treatment of lung diseases associated with total or partial AAT deficiency, primarily COPD.
ConclusionsAAT deficiency risk alleles causing COPD occur at a very low frequency among Mexican mestizo population. Although they are not directly linked in our study population with disease susceptibility, these risk alleles are associated with poorer lung function measurements. It is important to characterize how often these genetic risk variants occur in other Latin American populations.
FundingThis study was partially funded by the Consejo Nacional de Ciencia y Tecnología (CONACyT) of Mexico, code FOSIS S0008-87380.
Authors’ ContributionStudy design, and technical data by GPR, LOJV, RFV; statistical analysis and drafting of manuscript by GPR, LOJV, ARV, AC, RHS, FFT, JMRH, RFV.
Conflict of InterestsThe authors declare no conflict of interests.
Please cite this article as: Pérez-Rubio G, Jiménez-Valverde LO, Ramírez-Venegas A, Camarena Á, Sansores RH, Flores-Trujillo F, et al. Prevalencia de variantes de alto riesgo de alfa-1 antitripsina en población mestiza mexicana y su relación con los valores de la función pulmonar. Arch Bronconeumol. 2015;51:80–85.







