

Cystic fibrosis (CF) is the most frequent genetic disorder in the Caucasian race produced by the cystic fibrosis transmembrane conductance regulator (CFTR) gene disturbance. This gene, located on the long arm of chromosome 7, was first discovered by Andersen in 1938, although it was not until 1989 when it was identified as causing CF. This disease is transmitted in an autosomal recessive Mendelian inheritance pattern. Although it can be expressed phenotypically in different ways, it affects the exocrine epithelial cells of the respiratory system, the pancreas, the bile ducts, the sweat glands and the genitourinary system.1
In Spain, a decrease in the incidence of CF after the implantation of neonatal screening is recognized. However, the prevalence has increased2,3 in relation to the greater survival of these patients due to the improvement in the quality of care and the new therapeutic strategies making a disease that was initially considered pediatric4,5 become chronic.
The allelic heterogeneity of CFTR gene was described in the 1990s6 and it has recently been shown that the complexity of the mutation spectrum is greater than previously known.7
Alonso et al. managed to identify in Spain a total of 121 mutations of the CFTR gene, which represents 96% of the 1,954 Spanish alleles studied. Twelve of them presented frequencies higher than 1%, the most frequent being p.Phe508del (also known as F508del). Those that have a frequency lower than 0.5% are considered rare mutations7. To date, there is a global registry of CFTR mutations in the CF Mutation Database (CFTR1)8 and in the Clinical and Functional Translation of CFTR database (CFTR2) with constant updating,9 although some very rare and of uncertain clinical significance mutations continue to emerge. The identification of these gene disturbances and the phenotypic characterization of these patients allow us to know better the functional importance of the CFTR gene. Because of this, homozygous patients are required for these specific mutations, in which all the phenotypic expression can be attributed to that specific genetic alteration although unfortunately they are not usually prevalent.
We present the case of a 19-year-old Caucasian male homozygous for the p.Tyr1381X mutation (legacy name: Y1381X; DNAc: c.4143C>A). It is a mutation of synthesis defect (type I) whose functional alteration resulting is the absence of protein synthesis and characterized by the substitution of the amino acid tyrosine at position 1381 of CFTR by a termination codon.
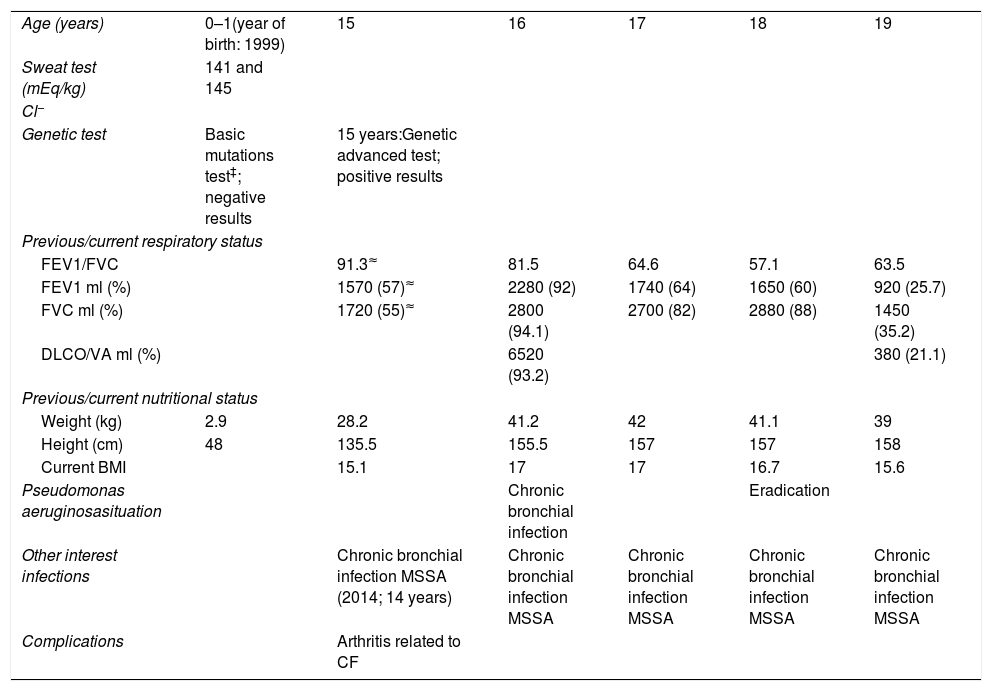
He was diagnosed with CF at 15 months old after admission for recurrent infections, diarrhoea and weight loss. The analysis of sweat test revealed that the concentration of chloride was 141 and 145mEq/L. The first genetic analysis performed on him after diagnosis was negative but it was used a basic panel of mutations. A most advanced genetic study was carried out at 2015 and characterized the real mutation of which up to that moment only one case had been registered in CFTR1.8 He was a full-term infant born before the CF neonatal screening implant. His birth weight was 2.9kg, birth length 48cm. Parental consanguinity was ruled out. His parents and his elder sister were carriers of this mutation but they were asymptomatic.
Until he was 15 years old the follow-up was performed by paediatrics. When he arrived at the Pulmonology Department he presented respiratory and digestive affectation, CF-associated arthritis and growth retardation. He presented chronic bronchial infection (CBI) by methicillin-sensitive Staphylococcus aureus (MSSA) since 14 years old without clinical repercussions. His treatment to date was: nebulized dornase alfa and hypertonic saline with hyaluronic acid (2.5mg/24h and 5ml/12h, respectively), budesonide/formoterol (160/4.5μg/12h), salbutamol (if need), azithromycin (250mg three times per week) and respiratory physiotherapy. He presented CBI by Pseudomonas aeruginosa (PA) since 16 years old so treatment was started with nebulized colistimethate sodium (1MIU/12h) and the PA infection was eradicated after two years. This therapy was discontinued in that moment. The patient did not have PA infections again; however, the CBI by MSSA persisted in time until the present.
Nowadays he presents a rapidly progressive clinical deterioration, highlighting recurrent infections. Last year he had a moderate exacerbation that was managed ambulatory with oral antibiotics and two severe exacerbations that required hospital admission due to respiratory failure. Due to respiratory deterioration, nebulized treatment with vancomycin was started for MSSA. Thoracic CT showed generalized cystic bronchiectasis. The respiratory deterioration has led to the request for evaluation of lung transplantation being currently on the waiting list.
Other comorbidities are severe pancreatic insufficiency (pancreatic gland atrophy and diffuse fatty replacement), iron-deficiency anaemia and acute severe malnutrition in the context of recent exacerbations.
The lung function, weight, height and CBI evolution is summarized in Table 1.
Main clinical features of the p.Tyr1381Y homozygous CF patient.
| Age (years) | 0–1(year of birth: 1999) | 15 | 16 | 17 | 18 | 19 |
| Sweat test (mEq/kg) | 141 and 145 | |||||
| Cl− | ||||||
| Genetic test | Basic mutations test‡; negative results | 15 years:Genetic advanced test; positive results | ||||
| Previous/current respiratory status | ||||||
| FEV1/FVC | 91.3≈ | 81.5 | 64.6 | 57.1 | 63.5 | |
| FEV1 ml (%) | 1570 (57)≈ | 2280 (92) | 1740 (64) | 1650 (60) | 920 (25.7) | |
| FVC ml (%) | 1720 (55)≈ | 2800 (94.1) | 2700 (82) | 2880 (88) | 1450 (35.2) | |
| DLCO/VA ml (%) | 6520 (93.2) | 380 (21.1) | ||||
| Previous/current nutritional status | ||||||
| Weight (kg) | 2.9 | 28.2 | 41.2 | 42 | 41.1 | 39 |
| Height (cm) | 48 | 135.5 | 155.5 | 157 | 157 | 158 |
| Current BMI | 15.1 | 17 | 17 | 16.7 | 15.6 | |
| Pseudomonas aeruginosasituation | Chronic bronchial infection | Eradication | ||||
| Other interest infections | Chronic bronchial infection MSSA (2014; 14 years) | Chronic bronchial infection MSSA | Chronic bronchial infection MSSA | Chronic bronchial infection MSSA | Chronic bronchial infection MSSA | |
| Complications | Arthritis related to CF | |||||
Despite genetic counselling, progress in the detection methods of mutations causing CF and implantation of neonatal screening in recent decades, there is still a small percentage of very rare mutations which are being identified and whose phenotypic behaviour is unknown. Its recognition allows us to advance in the study of this disease, especially in those cases of homozygous since its clinical behaviour is due solely to this mutation. Accordingly, this case report is an important step in this knowledge. It confirms once more the allelic richness of the Spanish population and the need to expand the range of CFTR mutation studies for those patient populations where mutations with standard mutation panels are not identified. It also suggests that the mutation p.Tyr1381X could be a severe mutation. Although in the last year the patient's deterioration has been rapidly progressive and also coincides with the appearance of a new chronic bronchial infection, we do not relate it to that infection due to not treating this germ of the most possibly pathogenic compared to others in the context of cystic fibrosis. It also seems to have respiratory and digestive involvement and rapidly progressive evolution, whose early detection and approach could improve the prognosis of these patients.







