




Information on the association of lung cancer (LC) and combined pulmonary fibrosis and emphysema (CPFE) is limited and derived almost exclusively from series in Asian populations. The main objective of the study was to assess the impact of LC on survival in CPFE patients and in patients with idiopathic pulmonary fibrosis (IPF).
MethodsA retrospective study was performed with data from patients with CPFE and IPF diagnosed in our hospital over a period of 5 years.
ResultsSixty-six patients were included, 29 with CPFE and 37 with IPF. Nine had a diagnosis of LC (6 with CPFE and 3 with IPF). Six patients (67%) received palliative treatment even though 3 of them were diagnosed atstage I–II. Overall mortality did not differ significantly between groups; however, in patients with LC, survival was significantly lower compared to those without LC (p=.044). The most frequent cause of death was respiratory failure secondary to pulmonary fibrosis exacerbation (44%). In a multivariate analysis, the odds ratio of death among patients with LC compared to patients without LC was 6.20 (p=.037, 95% confidence interval: 1.11–34.48).
ConclusionsLung cancer reduces survival in both entities. The diagnostic and therapeutic management of LC is hampered by the increased risk of complications after any treatment modality, even after palliative treatment.
La información sobre la asociación del cáncer de pulmón (CP) y combinación de fibrosis pulmonar y enfisema (CFPE) es limitada y procedente casi exclusivamente de series asiáticas. El objetivo principal del estudio fue valorar el impacto del CP en la supervivencia en la CFPE y en pacientes diagnosticados de fibrosis pulmonar idiopática (FPI).
MétodosSe realizó un estudio retrospectivo con los datos de pacientes con CFPE y FPI diagnosticados en nuestro centro en un periodo de 5 años.
ResultadosSe incluyó a 66 pacientes, 29 en el grupo de CFPE y 37 pacientes con FPI. Nueve tenían un diagnóstico de CP (6 con CFPE y 3 con FPI); 6 pacientes (67%) recibieron tratamiento paliativo a pesar de que 3 de ellos presentaban estadios i y ii. No hubo diferencias significativas en la mortalidad global de los 2 grupos; sin embargo, en los pacientes con CP la supervivencia fue significativamente menor con respecto a los que no tenían CP (p=,044). Las causas más frecuentes de muerte fue la insuficiencia respiratoria secundaria a la exacerbación de la fibrosis pulmonar (44%). En el análisis multivariante, la odds ratio de morir en los pacientes con CP respecto a los pacientes sin CP fue de 6,20 (p=,037, intervalo de confianza [IC] del 95%: 1,11 a 34,48).
ConclusiónEl CP empeora la supervivencia de estas 2 entidades. El manejo diagnóstico y terapéutico del CP se ve dificultado por el mayor riesgo de complicaciones posteriores al tratamiento elegido, incluso tras el tratamiento paliativo.
Combined pulmonary fibrosis and emphysema (CPFE) is a syndrome characterized by the coexistence of emphysema in the upper lobes and pulmonary fibrosis predominantly in the lower lobes, associated with a distinctive functional profile.1,2 Spirometric values appear to be practically unchanged, contrasting with a severe reduction in alveolar diffusing capacity (DLCO), hypoxemia, and desaturation on exertion. The most notable complications include pulmonary hypertension (PH)3,4 and lung cancer (LC), described in up to 47% of cases,5 both of which are determinant factors in patient survival.3–6
Information regarding the prevalence, characteristics, and prognosis of LC in CPFE is almost entirely derived from series in Asian populations.5,7–17 Recently, Girard et al.18 published the first description of the characteristics of LC in CPFE in a retrospective European cohort study conducted in France. In this study, poor prognosis was mainly attributed to limited therapeutic options in the management of these patients, due to their poor functional residual capacity and greater risk of complications after surgery or oncological treatment.
No studies have been performed in our setting to explore the relationship between LC and CPFE, so our aim was to analyze the influence of LC on survival in a series of CPFE patients. Clinical, functional and radiological data were compared with a series of patients with idiopathic pulmonary fibrosis (IPF), since this disease is also known to involve a greater risk of developing LC compared to the general population.19–21 Secondary objectives included a description of the prevalence and characteristics of LC in both groups.
MethodsThis was an observational, retrospective, cohort study of 2 patient groups (patients with a diagnosis of CPFE and patients with a diagnosis of IPF) recruited between December 2009 and December 2014. Patients were seen in the dedicated interstitial pulmonary disease (ILD) clinic of a third-level university hospital.
Patients with a diagnosis of CPFE were included using the criteria of Cottin et al.1 and Brillet et al.2: (a) evidence of areas of well-defined emphysema on high-resolution computed tomography (HRCT), defined as reduced attenuation compared with normal contiguous pulmonary parenchyma, delimited by a very thin wall (<1mm) or no wall, and/or multiple bullae (>1cm), predominantly in the upper lobes; the emphysematous lesions, described as a percentage of affected lung, had to be more than 10%; and (b) presence of DILD with significant characteristics of pulmonary fibrosis, defined as reticular pulmonary opacities predominantly in the periphery and bases, honeycombing, distorted lung architecture and/or retraction, bronchiectasis, or bronchiolectasis. Opacities and/or focal areas of ground glass alveolar consolidation may be associated with this pattern, but must not be prominent. The diagnosis of IPF was made on the basis of the SEPAR diagnostic and therapeutic guidelines for IPF,22 which establish that the definitive diagnosis of IPF requires: (a) exclusion of other defined clinical entities or diffuse pulmonary parenchymal diseases of known etiology (environmental or occupational exposure, connective tissue diseases, drug toxicity), and (b) presence of a histological pattern of usual interstitial pneumonia (UIP) in the study of lung tissue obtained by surgical lung biopsy, or else radiological evidence of the UIP pattern on HRCT, or both. Cases with inconclusive diagnoses were reviewed by the ILD multidisciplinary committee of our hospital (Fig. 1).

Echocardiography data were collected from patients with a clinical or radiological suspicion of PH. The arbitrary criteria proposed by Galiè et al.23 were used to evaluate the presence of PH. In patients with a diagnosis of LC, initial symptoms, fiberoptic bronchoscopy (FB) findings, staging, histology, treatment and months of survival after diagnosis were collected. All patients were initially staged according to the seventh edition of the TNM classification,24 with pathological staging according to the World Health Organization classification.25
Acute exacerbation of pulmonary fibrosis was defined as respiratory deterioration with no identifiable cause, according to the criteria proposed by Collard et al.26
For analysis of mortality, the following data were recorded: (a) date of diagnosis of CPFE and IPF, (b) date of diagnosis of LC, and (c) date of death. The latter was confirmed from digital clinical records and/or the hospital death records. Lung function tests and the 6-minute walk test (6MWT) were performed according to international recommendations.27–30 The composite physiologic index (CPI) was calculated using the following formula: 91−(0.65×percentage predicted DLCO)–(0.53×percentage predicted forced vital capacity [FVC])+(0.34×percentage predicted forced expiratory volume in 1 second [FEV1]).31
Statistical AnalysisData were analyzed using the Stata statistical package, version 13 (StataCorp LP, College Station, USA). All data were tabulated as mean and standard deviation for quantitative variables and as absolute numbers and percentages for qualitative variables. Survival curves for the groups were constructed according to the Kaplan–Meier methods and compared using the log-rank test.
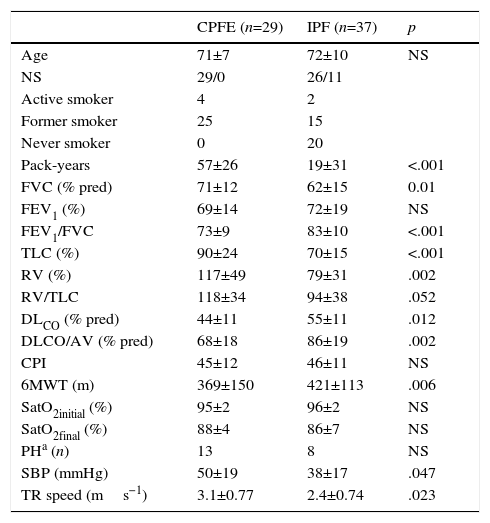
ResultsA total of 66 patients were included, 29 in the CPFE group and 37 in the IPF group. Median follow-up was 24.3 months (interquartile range 11.4–42.5). All CPFE patients were men and had a significantly higher accumulated pack-year index than patients with IPF (p<.001). Patients with CPFE had significantly higher values in the following functional parameters (expressed as percentage of predicted value): FVC, ratio of forced expiratory volume in 1 second (FEV1) and FVC (FEV1/FVC), total lung capacity (TLC), and residual volume. DLCO and DLCO/alveolar volume ratio were also lower compared to patients with IPF (p<.05). PH estimated by echocardiography was more common in patients with CPFE (45% vs 22% in the IPF group). These patients had higher systolic blood pressure, and a greater tricuspid regurgitation velocity compared to patients with IPF (p=.047 and .023, respectively). These data are summarized in Table 1.
Demographic and Functional Characteristics of Patients.
| CPFE (n=29) | IPF (n=37) | p | |
|---|---|---|---|
| Age | 71±7 | 72±10 | NS |
| NS | 29/0 | 26/11 | |
| Active smoker | 4 | 2 | |
| Former smoker | 25 | 15 | |
| Never smoker | 0 | 20 | |
| Pack-years | 57±26 | 19±31 | <.001 |
| FVC (% pred) | 71±12 | 62±15 | 0.01 |
| FEV1 (%) | 69±14 | 72±19 | NS |
| FEV1/FVC | 73±9 | 83±10 | <.001 |
| TLC (%) | 90±24 | 70±15 | <.001 |
| RV (%) | 117±49 | 79±31 | .002 |
| RV/TLC | 118±34 | 94±38 | .052 |
| DLCO (% pred) | 44±11 | 55±11 | .012 |
| DLCO/AV (% pred) | 68±18 | 86±19 | .002 |
| CPI | 45±12 | 46±11 | NS |
| 6MWT (m) | 369±150 | 421±113 | .006 |
| SatO2initial (%) | 95±2 | 96±2 | NS |
| SatO2final (%) | 88±4 | 86±7 | NS |
| PHa (n) | 13 | 8 | NS |
| SBP (mmHg) | 50±19 | 38±17 | .047 |
| TR speed (ms−1) | 3.1±0.77 | 2.4±0.74 | .023 |
6MWT: 6-minute walk test; % pred: percentage of predicted value; AV: alveolar volume; CPFE: combined pulmonary fibrosis and emphysema; CPI: composite physiologic index; DLCO: diffusing capacity of the lung for carbon monoxide; FEV1: forced expiratory volume in 1 second; FEV1/FVC: forced expiratory volume in 1 second and forced vital capacity ratio expressed as an absolute percentage; FVC: forced vital capacity; IPF: idiopathic pulmonary fibrosis; PH: pulmonary hypertension; RV: residual volume; RV/TLC: ratio of RV and total lung capacity; SPB: systolic blood pressure; SatO2final: oxygen saturation at end of 6MWT; SatO2initial: oxygen saturation at start of 6MWT; TLC: total lung capacity; TR: tricuspid regurgitation.
Data are presented as mean±SD.
CPFE and IPF diagnosis preceded LC diagnosis by a mean of 9.8 months in all patients. Nine patients had a diagnosis of LC (13.6%); 6 of them (66.6%) had CPFE. All patients in this group were men with a history of smoking. The prevalence of LC in this series was 20% among patients with CPFE and 8% among those with IPF.
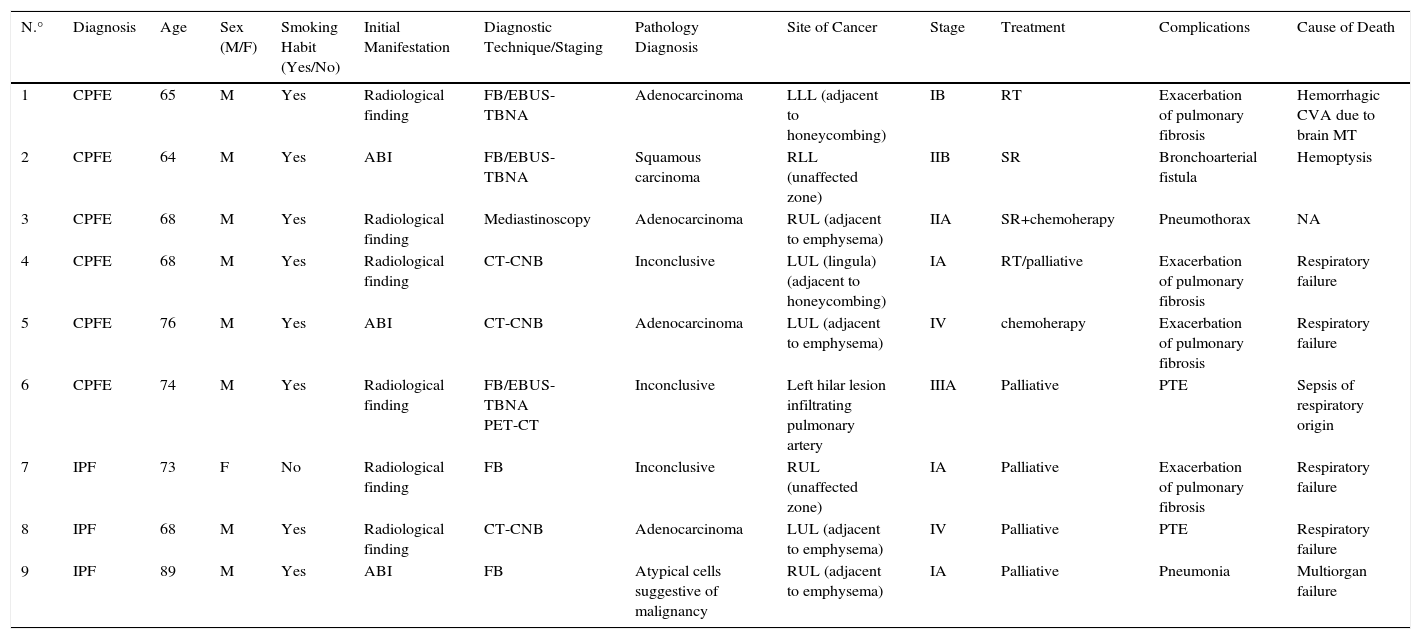
The initial suspicion of LC in all patients was radiological, detected in routine follow-up HRCTs (67%). Three patients reported an increase in habitual cough and had a diagnosis of acute bronchial infection (33%). The tumor was visible on the chest radiograph of 5 of the 9 patients (55%). In 2 cases (22%), the tumor lesion was situated adjacent to areas of pulmonary fibrosis (Fig. 2). Most tumors were located in upper lobes, and 5 were within the areas of emphysema (56%). The most widely used diagnostic strategy was FB, followed by real-time endobronchial ultrasound with transbronchial needle aspiration (EBUS-TBNA) and computed tomography-guided core needle biopsy (CT-CNB). A pathology diagnosis was not obtained from 3 patients (33%). The most common histological subtype was adenocarcinoma: 6 patients had localized stages (I–II) at the time of diagnosis (67%). Only 2 patients received surgical treatment, one of whom died on day 17 post-surgery (bilobectomy). The other underwent lobectomy, followed by adjuvant chemotherapy, and has survived 39 months to date. The most common treatment modality was palliative care. Most commonly observed complications were exacerbations of pulmonary fibrosis in 4 patients (44%) (Table 2).
 Chest radiograph (posteroanterior projection): Poorly defined nodular focal pulmonary opacity (arrow) is observed in the base of the left lung, and a reticular interstitial involvement in both lung bases. (B) HRCT showing presence of emphysema in the upper lobes. (C) Chest HRCT confirming presence of a pulmonary mass with lobulated and spiculated borders (arrow head) in the lingula, adjacent to an area of honeycombing. CPFE: combined pulmonary fibrosis and emphysema; LC: lung cancer; HRCT: high-resolution computed tomography.")
Patient with CPFE and LC. (A) Chest radiograph (posteroanterior projection): Poorly defined nodular focal pulmonary opacity (arrow) is observed in the base of the left lung, and a reticular interstitial involvement in both lung bases. (B) HRCT showing presence of emphysema in the upper lobes. (C) Chest HRCT confirming presence of a pulmonary mass with lobulated and spiculated borders (arrow head) in the lingula, adjacent to an area of honeycombing. CPFE: combined pulmonary fibrosis and emphysema; LC: lung cancer; HRCT: high-resolution computed tomography.
Characteristics, Treatment and Progress of Patients with Lung Cancer.
| N.° | Diagnosis | Age | Sex (M/F) | Smoking Habit (Yes/No) | Initial Manifestation | Diagnostic Technique/Staging | Pathology Diagnosis | Site of Cancer | Stage | Treatment | Complications | Cause of Death |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | CPFE | 65 | M | Yes | Radiological finding | FB/EBUS-TBNA | Adenocarcinoma | LLL (adjacent to honeycombing) | IB | RT | Exacerbation of pulmonary fibrosis | Hemorrhagic CVA due to brain MT |
| 2 | CPFE | 64 | M | Yes | ABI | FB/EBUS-TBNA | Squamous carcinoma | RLL (unaffected zone) | IIB | SR | Bronchoarterial fistula | Hemoptysis |
| 3 | CPFE | 68 | M | Yes | Radiological finding | Mediastinoscopy | Adenocarcinoma | RUL (adjacent to emphysema) | IIA | SR+chemoherapy | Pneumothorax | NA |
| 4 | CPFE | 68 | M | Yes | Radiological finding | CT-CNB | Inconclusive | LUL (lingula) (adjacent to honeycombing) | IA | RT/palliative | Exacerbation of pulmonary fibrosis | Respiratory failure |
| 5 | CPFE | 76 | M | Yes | ABI | CT-CNB | Adenocarcinoma | LUL (adjacent to emphysema) | IV | chemoherapy | Exacerbation of pulmonary fibrosis | Respiratory failure |
| 6 | CPFE | 74 | M | Yes | Radiological finding | FB/EBUS-TBNA PET-CT | Inconclusive | Left hilar lesion infiltrating pulmonary artery | IIIA | Palliative | PTE | Sepsis of respiratory origin |
| 7 | IPF | 73 | F | No | Radiological finding | FB | Inconclusive | RUL (unaffected zone) | IA | Palliative | Exacerbation of pulmonary fibrosis | Respiratory failure |
| 8 | IPF | 68 | M | Yes | Radiological finding | CT-CNB | Adenocarcinoma | LUL (adjacent to emphysema) | IV | Palliative | PTE | Respiratory failure |
| 9 | IPF | 89 | M | Yes | ABI | FB | Atypical cells suggestive of malignancy | RUL (adjacent to emphysema) | IA | Palliative | Pneumonia | Multiorgan failure |
ABI: acute bronchial infection; CVA: cerebrovascular accident; CPFE: combined pulmonary fibrosis and emphysema; chemoherapy: chemotherapy; CT-CNB: computed tomography-guided core needle biopsy; EBUS-TBNA: real-time endobronchial ultrasound-transbronchial needle aspiration; FB: fiberoptic bronchoscopy; IPF: idiopathic pulmonary fibrosis; LLL: left lower lobe; LUL: left upper lobe; M/F: male/female; MT: metastasis; NA: not applicable; PET-CT: positron emission tomography-computed tomography; PTE: pulmonary thromboembolism; RLL: right lower lobe; RUL: right lower lobe; RT: radiation therapy; SR: surgical resection.
The survival curve did not reveal any differences between CPFE patients compared to IPF patients (p=.10) (Fig. 3). Twelve-month survival in patients with LC was 0.78 (95% confidence interval [CI]: 0.36–0.94). Survival at 24 months was 0.67 (95% CI: 0.28–0.88). Survival at 36 months was 0.13 (95% CI: 0.01–0.44). In patients without LC, accumulated survival at 12, 24, 36, and 48 months was 0.85 (95% CI: 0.73–0.92), 0.74 (95% CI: 0.59–0.84), 0.59 (95% CI: 0.43–0.73), and 0.49 (95% CI: 0.32–0.64), respectively. The difference in survival between the 2 groups was statistically significant (p=.044) (Fig. 4). In the multivariate analysis, the odds ratio (OR) of death among patients with LC compared to patients without LC was 6.20 (p=.037, 95% CI: 1.11–34.48).


Our data show that LC significantly reduces survival in patients with CPFE and IPF. The risk of death with this complication is up to 6 times greater compared to patients without LC. Lack of success in diagnosing the histological type in a third of cases and the high rate of complications after the selected treatment modality underline the difficulties in the diagnosis and management of LC in this group of patients.
To our knowledge, this is the first study to describe a series of CPFE patients in Spain. Data on the prognosis of LC in CPFE are mostly derived from Asian series, and these results need to be replicated in other ethnic groups.6 In contrast, the influence of LC on IPF has been widely studied in different populations, and identified as an independent factor of death in this entity.19–21,32–34 Girard et al.18 performed a study in 47 patients with CPFE: in 9 (19%) of these patients a diagnosis of LC could not be obtained. Twenty patients (42%) could not be given the cancer treatment recommended in the international guidelines due to their functional characteristics.
In our series, CPFE patients met the characteristics described above, and their functional profile was specific and different from that of patients with IPF.1,5,6 They were mainly men, with a history of smoking and a higher pack-year index that the IPF group. The cohorts studied in the literature are mostly men, but the significance of this finding remains unclear.6,35 They had higher than predicted percentages of FVC and TLC, and, in contrast, profound changes in DLCO. In CPFE, PH is a cardiovascular complication associated with poorer prognosis.3,4 In line with previous studies,1,3,4 PH estimated by echocardiography in our patients was more common in the CPFE group.
It is sometimes difficult to distinguish between these 2 entities in daily clinical practice, and it is essential for cases to be discussed by a ILD multidisciplinary team before reaching a differential diagnosis. The CPI has been shown to have a better predictive value than individual functional parameters in the follow-up of IPF patients6,31; however, in our study, we did not find this measurement useful for differentiation. It would be useful to have studies analyzing new functional or radiological indices that might reflect the degree of functional and involvement that characterize CPFE patients.
In our study, the prevalence of LC was higher in the CPFE group (20% vs 8% in IPF patients); however, due to the small sample size, this result must be interpreted with caution. Kwak et al.12 found a significantly higher risk of LC among CPFE patients than in patients with isolated emphysema, but they did not find significant differences in patients with IPF. Reports in the literature show that 1 in 10 patients with IPF develop LC, with a variable prevalence, reaching up to 48% in autopsy studies.32 Some authors consider that the greatest risk of LC in CPFE patients is due to the combined “triple effect” in these patients of smoking, emphysema, and pulmonary fibrosis.6 Cigarette smoke is the main causative agent of LC (90% of cases),36 while emphysema has been shown to be an independent risk factor for the development of this entity.36,37 In line with this observation, though at odds with other studies,12,32 we found that lesions were located in areas of emphysema in almost 2 thirds of LC cases. In this respect, the information on the tumor site is limited in published series.12 Most tumors were located in the upper lobes, consistent with the western cohort described by Girard et al.18
Although both entities represent a risk group for the development of LC, current clinical guidelines contain no recommendations on screening strategies in these diseases.22,36 The lack of diagnosis in one third of our cases shows the difficulty of using invasive procedures in patients with a high risk of complications, given their parenchymal changes.18
In non-small cell LC stages I and II, radical surgery offers the best possibility of cure.36 However, several recent studies have indicated a poorer prognosis in both CPFE and IPF patients after surgical resection, due to a higher rate of post-operative complications (particularly acute exacerbation of pulmonary fibrosis and early LC relapse).14,21,32,34 In our series, only 2 patients were receiving surgical treatment, so it is difficult to extract conclusions in this respect. However, the patient who has survived, disease-free, until the time of writing underwent lobectomy, in contrast to the other patient who died in the late post-operative period (after bilobectomy). This observation is in line with previous studies that supported more conservative surgical interventions in these patients, such as segmentectomy or lobectomy.21,34,38
Although 67% of LC patients were in stages I–II, the most widely used treatment modality was palliative care. Treatment with chemotherapy or radiation therapy (2 cases with both treatments) was also an ineffective option. The most common complication in LC patients was exacerbation of pulmonary fibrosis, an event commonly described in the published series.13,34,38
In this study, LC significantly reduced patient survival and was associated with a 6-fold increase in mortality rate compared to patients without LC. In accordance with our findings, Tomassetti et al.34 reported a hazard ratio of 7 in a cohort of 186 patients with IPF. In another recent study, LC was a significant predictor of death in the “comorbidome” of patients with IPF.33
CPFE per se has been identified by some authors as an independent prognostic factor for LC relapse and survival.14 Girard et al.18 confirmed the unfavorable prognosis of LC in these patients, in line with our results and with those of the Asian cohort studies.
As for overall mortality, we found no significant differences between patients with CPFE and IPF, a finding that coincides with those of Jankowich and Rounds39 in a study with a very similar sample and follow-up to ours. In this respect, most studies available to date confirm that the already precarious survival in both entities is substantially affected by extrapulmonary comorbidities (cardiovascular disease, including PH, gastroesophageal reflux, and LC3,4,6,9–21,32–34). Newly approved drugs for the treatment of IPF (pirfenidone and nintedanib) offer new possibilities in this respect, and may help modify the natural disease course of this entity.40 Future studies will be needed to compare historical cohorts and analyze the impact of these treatments on survival and incidence of comorbidities.
We recognize that our study has some limitations that prevent generalization of our results. Firstly, it was carried out in a single center and its retrospective design may introduce limitations in data analysis. Since the primary objective of this study was to analyze the impact of LC on mortality of CPFE and IPF patients for the first time in a Spanish series, our findings answer the research question. Secondly, we cannot establish the influence of new, recently approved antifibrotic treatments on the incidence of LC or on the overall survival of the series. We believe that this fact does not affect the results presented here, given the low number of treated patients included in the sample at the time of recruitment. Finally, 2 relatively small patient groups were studied, with the consequent limitation on comparisons and the extrapolation of results. However, we believe that our sample is representative of patients treated in real-world situations in third-level hospitals in our setting. Investigational collaboration and national multicenter registries are needed to generate more information on this topic.
In conclusion, our results show that in patients with CPFE and IPF, LC is a complication that demonstratively increases mortality. These patients are more vulnerable to developing complications related with the selected anticancer treatment, even after palliative management. These considerations must be taken into account in clinical guidelines, in order to help standardize the diagnostic and therapeutic approach to LC in these patients.
FundingKarina Portillo Carroz received a research grant from the Spanish Society of Pulmonology and Thoracic Surgery (2013) and an “EPID-futuro” grant sponsored by Roche (2013).
Conflict of InterestsThe authors state that they have no conflict of interests.
Please cite this article as: Portillo K, Perez-Rodas N, García-Olivé I, Guasch-Arriaga I, Centeno C, Serra P, et al. Cáncer de pulmón en pacientes con combinación de fibrosis pulmonar y enfisema y fibrosis pulmonar idiopática. Estudio descriptivo en una serie española. Arch Bronconeumol. 2017;53:304–310.







