

La linfangioleiomiomatosis es una enfermedad rara caracterizada por quistes pulmonares con proliferación anómala del sistema linfático. Se presenta casi en exclusiva en mujeres en edad fértil debido a una influencia hormonal, por lo que resulta sumamente infrecuente en pacientes posmenopáusicas. En estos casos suele tener relación con tratamientos hormonales sustitutivos. Es conocido que esta enfermedad tiene un alto grado de asociación con otras afecciones como la esclerosis tuberosa y los angiomiolipomas renales.
Presentamos el caso de una paciente posmenopáusica afecta de linfangioleiomiomatosis, sin ningún antecedente de tratamientos hormonales, en la que se detectaron, además, angiomiolipomas renales y criterios clínicos indicativos de esclerosis tuberosa en grado de probable.
Lymphangioleiomyomatosis is a rare disease characterised by pulmonary cysts with abnormal proliferation in the lymphatic system. It occurs almost exclusively in women of fertile age due to a hormonal influence, for this reason it is extremely rare in post-menopausal patients. In these cases it is usually associated to hormone replacement therapies. It is known that this diseases is strongly associated with other conditions, such as tuberous sclerosis and renal angiomyolipomas.
We present a case of a post-menopausal patient suffering from lymphangioleiomyomatosis, with no history of hormone therapy, in whom were also detected renal angiomyolipomas and clinical signs indicative of a probable tuberous sclerosis.
La linfangioleiomiomatosis (LAM) es una entidad infrecuente de etiología desconocida, que afecta casi exclusivamente a mujeres en edad fértil. En una serie del año 2000 en nuestro país se estimaba una prevalencia de 1 caso por 1.000.000 de habitantes1. Aparece como una enfermedad aislada o en asociación con esclerosis tuberosa, en la que un 2–3% de los pacientes puede presentar LAM2. Sin embargo, si se realiza un cribado específico en mujeres con esclerosis tuberosa, se llega a detectar hasta un 40% de LAM, lo cual indica que muchas de estas pacientes no desarrollan enfermedad pulmonar clínica3. La LAM se caracteriza por la destrucción quística del parénquima pulmonar, asociada a una proliferación incontrolada de células musculares lisas atípicas, denominadas células LAM3. La afectación predominante es la pulmonar, aunque no es infrecuente encontrar afección extratorácica. Por otra parte, es sabido que los angiomiolipomas renales se detectan en un 40–80% de los pacientes con esclerosis tuberosa4,5; sin embargo, en pacientes con LAM también hay una asociación importante (15–30%), lo que apunta a la existencia de similitudes entre ambas enfermedades4,6.
Presentamos un caso de LAM en una mujer posmenopáusica con esclerosis tuberosa en grado de probable según los criterios de la Tuberous Sclerosis Alliance7 y angiomiolipomas renales.
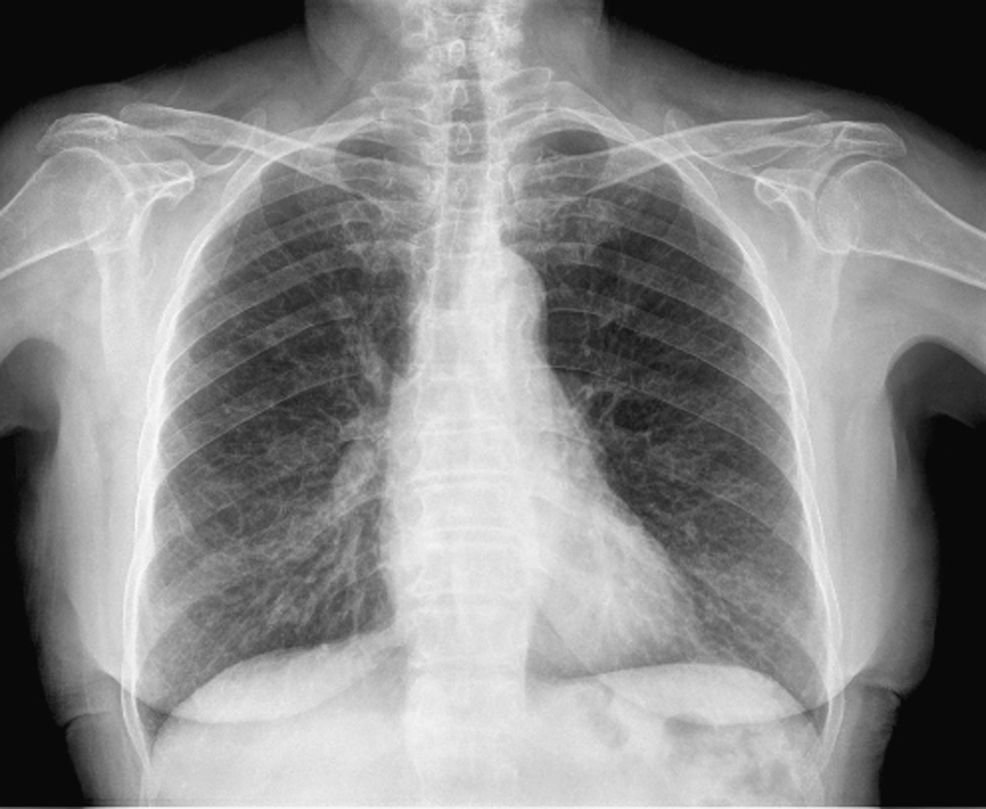
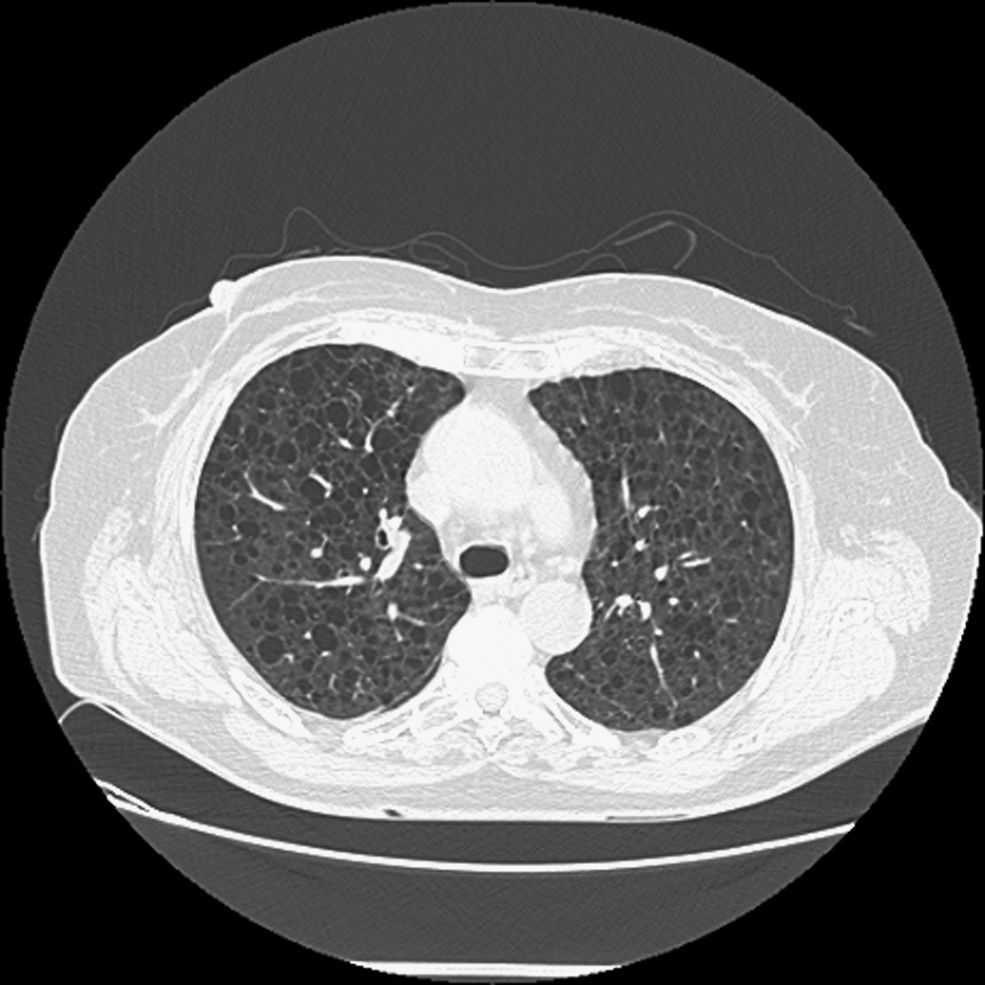
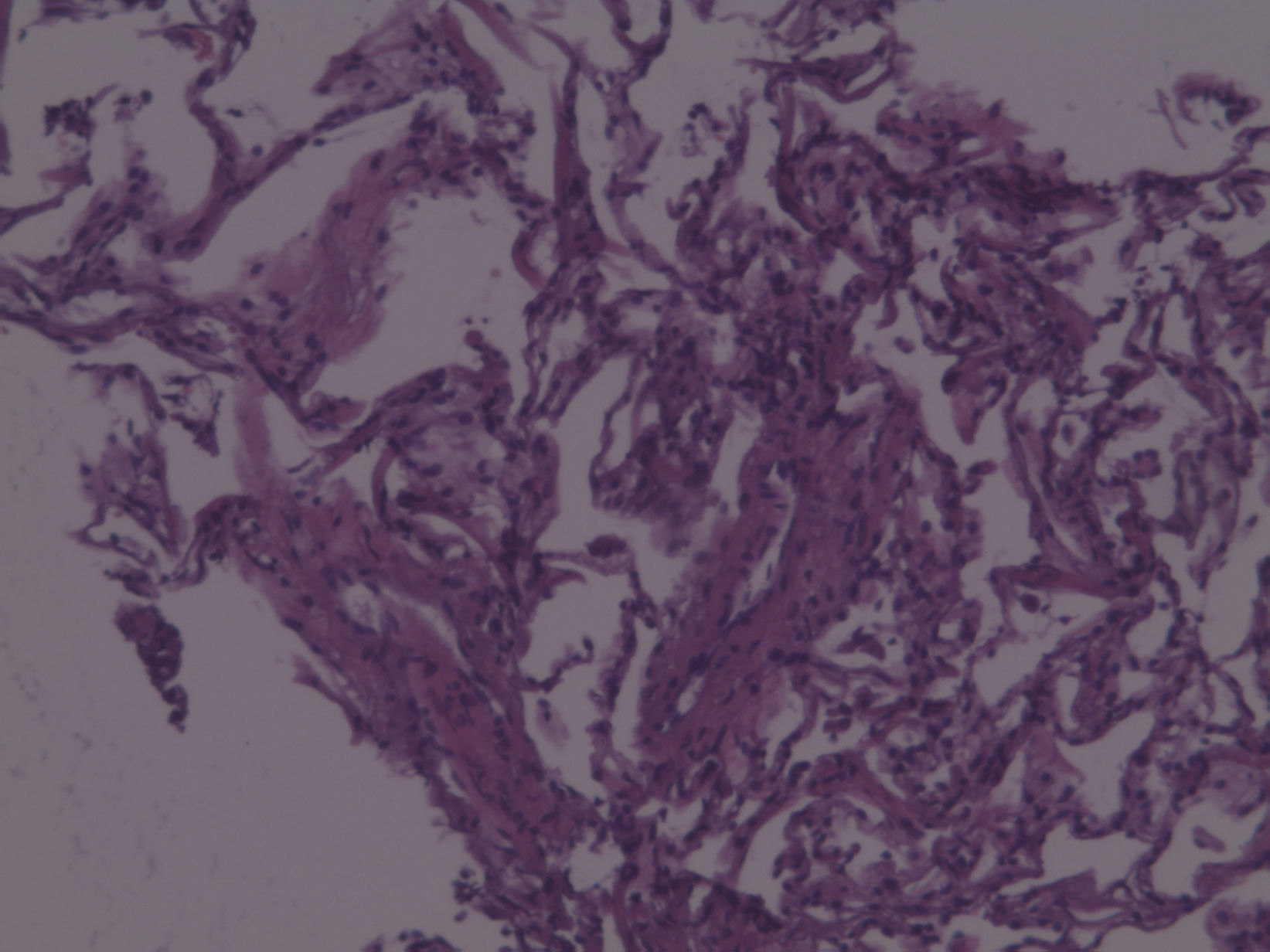
Observación clínicaMujer de 70 años, no fumadora, que consultó al Servicio de Medicina Interna por disnea de esfuerzo. Entre sus antecedentes patológicos destacaban un episodio de neumonía hacía 20 años y un tumor cerebral intervenido y tratado con radioterapia 3 años antes en otro hospital, del que desconocemos la histología, y del que le había quedado como secuela una parálisis facial izquierda. La exploración física fue normal, con saturación de oxígeno del 98%. Únicamente en la región periorbitaria destacaban unas discretas lesiones cutáneas indicativas de angiofibromas, sin detectarse lesiones en ninguna otra región corporal. En la radiografía de tórax (fig. 1) se observó un patrón intersticial, y la tomografía computarizada (fig. 2) evidenció múltiples imágenes quísticas en todo el campo pulmonar de ambos hemitórax, de morfología redonda y pared fina, sin estructuras vasculares en su interior ni engrosamiento de los septos. En la exploración funcional respiratoria la capacidad vital forzada (FVC) era de 1.970ml (un 68% del valor teórico), el volumen espiratorio forzado en el primer segundo (FEV1) de 1.260ml (un 61% del valor teórico) y FEV1/FVC de 63,65%. Se practicó una fibrobroncoscopia con lavado bronquioalveolar y biopsias transbronquiales. El estudio anatomopatológico (fig. 3) de las biopsias estableció el diagnóstico de linfangiomiomatosis pulmonar. Se llevó a cabo un estudio de receptores hormonales, que mostró positividad leve para los receptores de progesterona en numerosas células alrededor de las estructuras vasculares. A fin de descartar tumoraciones asociadas se efectuaron ecografía y tomografía computarizada abdominales, lo que permitió descubrir una lesión de densidad grasa en el polo inferior del riñón derecho, de 20mm, indicativa de angiomiolipoma, y 2 lesiones de características semejantes pero de menor tamaño en el polo inferior (7mm) y tercio medio (7,8mm) del riñón izquierdo.



La LAM es una enfermedad pulmonar rara, de etiología y patogenia aún desconocidas. Sabemos que la enfermedad progresa lentamente hacia la muerte en 8–10 años8 y que se manifiesta en etapas fértiles de la vida de la mujer, con exacerbaciones durante la menstruación o los embarazos, lo que indica una probable influencia hormonal. Diversos estudios señalan que la progresión de la LAM puede verse acelerada por los estrógenos y enlentecida por la progesterona9–11. Está documentado que en las biopsias pulmonares pueden hallarse receptores de esteroides, estrógenos y progestágenos11. La enfermedad es extremadamente infrecuente en mujeres posmenopáusicas, y en estos casos se relaciona casi exclusivamente con tratamientos estrogénicos sustitutivos12,13. Sin embargo, el hallazgo de LAM en preadolescentes, posmenopáusicas e incluso en varones indica que los estrógenos no son el único factor etiológico.
En el caso que nos ocupa, se trata de una paciente posmenopáusica sin antecedentes de tratamiento hormonal sustitutivo, que podría tener una esclerosis tuberosa de base (diagnóstico en grado de probable)7 como único factor predisponente. La mayoría de los autores propone que la asociación de angiomiolipomas con LAM pulmonar es una forma “frustrada” de esclerosis tuberosa, y teorizan que LAM y la esclerosis tuberosa representan una gama de una misma enfermedad4,14. El solapamiento entre estas 3 enfermedades (LAM, esclerosis tuberosa y angiomiolipoma renal) se denomina complejo esclerosis tuberosa15.
En las mujeres mayores la enfermedad se presenta con clínica similar a la de las jóvenes, con la excepción de que el curso clínico es más largo y más benigno después de la menopausia.
Los tratamientos actuales para la LAM se basan principalmente en el antagonismo de la acción de los estrógenos9–11,13, siendo los más comúnmente empleados los tratamientos con progestágenos, el tamoxifeno o la castración quirúrgica por ooforectomía. La rapamicina es un agente inmunodepresor con indicación para el tratamiento de la esclerosis tuberosa, y con ella se ha constatado una ligera disminución del tamaño de los angiomiolipomas renales, aunque en algunos casos éstos tienen tendencia a aumentar de nuevo de volumen al retirar el fármaco. En cambio, en los pacientes con LAM con quienes se usó la rapamicina se detectó una mejoría en la espirometría de forma mantenida tras la interrupción del tratamiento16. En última instancia, en los casos graves con afectación progresiva de la función respiratoria se debe considerar el trasplante pulmonar.







