



We present the case of a 43-year-old male who is an ex-smoker. He presented progressive dyspnea and hypoxia, and chest radiograph showed bilateral diffuse interstitial alterations. Thoracic CT revealed worsened bilateral ground glass opacities, with no signs of focal consolidation or interstitial alterations (Fig. 1a). The patient was intubated and ventilated and a lung biopsy was taken. The biopsy demonstrated a combination of interstitial lymphocytic infiltrate with altered architecture in many areas, associated with elements of bronchiolitis obliterans that mainly affected the airways at the level of the alveolar ducts, but in a few areas it also affected the respiratory bronchioles. This combination is described as bronchiolitis interstitial pneumonitis (BIP) (Fig. 1b). The patient improved slowly, with no specific treatment. At the follow-up consultation six months later, there was evidence of a complete recuperation. A repeat thoracic CT showed complete resolution of the BIP.
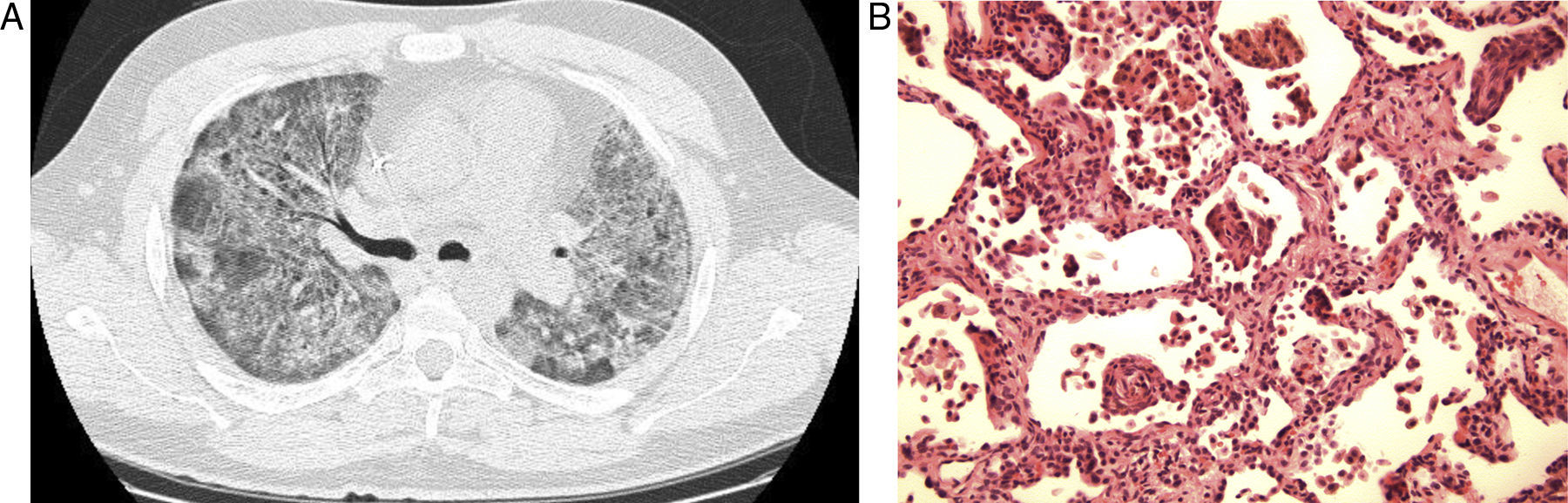
The classification of interstitial pneumopathies is largely based on the patterns observed in open or transbronchial biopsy samples. In 2008, Mark et al. described a cohort of 31 patients who, upon anatomopathological examination, presented interstitial pneumonitis with bronchiolitis.1 100% of the cases reported in the only series published presented signs of bronchiolitis obliterans as well as interstitial fibrosis. It should be noted that the fibrosis was at a distance from the bronchiolar disease, which suggests that the fibrosis and the bronchiolar disease are not a direct consequence of one another. In all the cases, the interstitial fibrosis was observed at a greater rate than the lymphocytic interstitial inflammation.
Interstitial pneumonitis was defined as an interstitial infiltrate of lymphocytes and fibrosis.1 The inflammation and fibrosis were present in all the cases. Interstitial pneumonitis with bronchiolitis presented a response to corticosteroids that was inferior to that of bronchiolitis obliterans organizing pneumonia (BOOP), but superior to that of usual interstitial pneumonitis (UIP) and non-specific interstitial pneumonia/fibrosis (NSIP). Fibroblastic foci were observed in only 21% of the cases.1
Another important differential diagnosis that should also be taken into account in this case is respiratory bronchiolitis-associated interstitial lung disease (RB-ILD). RB-ILD is often observed in patients who are current or ex-smokers, although its appearance has also been described in non-smokers.2 Evidence has been obtained indicating an accumulation of macrophages with dark pigmentation in the respiratory bronchioles and in the surrounding airspace2 associated with a submucosal and peribronchiolar infiltration dotted with lymphocytes and histiocytes. Peribronchiolar fibrosis may also be observed.3 In this disorder, fibroblastic foci are not observed, which differentiates it from other idiopathic interstitial pneumonias.3
This case poses the question of the utility of open lung biopsies being more frequently done4 and the need for a more sophisticated histologic analysis. The anatomopathological examination is less useful when obtained later on in the course of the disease or after treatment is initiated.5
Our case is a contribution towards the limited amount of data published to date about this entity, which is reported very infrequently. It is the first reported case of spontaneous resolution of BIP. It is very important to be more aware of this entity, as many cases may not be diagnosed or may currently be misdiagnosed.
Please cite this article as: Chong SG, et al. Neumonitis intersticial con bronquiolitis: presentación de un caso y revisión de la literatura. Arch Bronconeumol. 2012;48:262–3.







