






Las hemorragias alveolares difusas son cuadros clínicos que pueden ser catastróficos si no se diagnostican y tratan a tiempo. Suelen estar causadas en gran parte por vasculitis de vasos pequeños pulmonares. Existen 3 grandes grupos: a) las pauciinmunitarias, generalmente asociadas a capilaritis y anticuerpos citoplásmicos antineutrófilos; b) las producidas por depósitos inmunológicos, que pueden detectarse mediante inmunofluorescencia, y c) un gran grupo misceláneo, que incluye toxicidad por fármacos, infecciones y causas idiopáticas. El diagnóstico se basa en la integración de signos, síntomas, estudios serológicos y morfológicos. Se debe recomendar la realización de una biopsia por videotoracoscopia en los pacientes con hemorragia alveolar difusa de causa inexplicada, sin un diagnóstico previo de enfermedad sistémica, en la que los estudios serológicos no proporcionan datos concluyentes, y en general en aquellos pacientes con un elevado índice de sospecha de que estén desarrollando una hemorragia alveolar difusa. En todos estos casos, la biopsia debe remitirse en fresco a los servicios de anatomía patológica para permitir la congelación de un fragmento tisular, que será utilizado para el estudio por inmunofluorescencia.
Diffuse alveolar hemorrhage is a clinical syndrome that can be life threatening if not diagnosed and treated in time. In most cases it occurs largely as a result of small-vessel vasculitis of the lungs. The many different forms can be classified into 3 large groups: a) pauciimmune disease, which generally involves pulmonary capillaritis and is associated with the presence of antineutrophil cytoplasmic antibodies; b) syndromes caused by immune deposits, which can be detected by immunofluorescence; and c) a large miscellaneous group that includes drug reactions, infections, and idiopathic disease. Diagnosis is based on a combination of signs, symptoms, serology, and histology. Biopsy with video-assisted thoracoscopy should be recommended in patients with diffuse alveolar hemorrhage without known cause and with no prior diagnosis of systemic disease, in whom serology studies do not reveal conclusive data, and in general in those patients for whom there is a high level of suspicion of diffuse alveolar hemorrhage. In all such cases, the fresh biopsy material should be sent to the pathology laboratory for preparation of frozen sections to be used for immunofluorescence.
El cuadro clínico de la hemorragia alveolar es un conjunto sindrómico que tiene entidad como tal, que generalmente se asocia a situaciones de gravedad y que merece ser tratado de forma detallada1. Sin embargo, el enfoque diagnóstico suele ser dificultoso, ya que, aunque una parte de los pacientes se presenta con el diagnóstico previo de vasculitis o colagenosis y enfermedad renal asociada, y los hallazgos clínicos clásicos de infiltrados alveolares bilaterales, hemoptisis, caída de los valores de hemoglobina y/o hematocrito y aumento de la capacidad de difusión del monóxido de carbono por encima del 30%, es relativamente frecuente que una hemorragia alveolar con capilaritis necrosante sea la forma de inicio de una enfermedad sistémica, que falten muchos de los signos clásicos o que el síndrome se encuentre limitado a los pulmones. Es en estos casos de diagnóstico difícil donde se deben buscar exhaustivamente los signos de enfermedad sistémica (sinusitis, vasculitis leucocitoclástica cutánea, iridociclitis, sinovitis y glomerulonefritis) y donde la biopsia pulmonar puede aportar datos significativos.
La enfermedad inflamatoria de los vasos sanguíneos ha sido desde siempre un campo de difícil diagnóstico y manejo. Los precursores en el diagnóstico y reconocimiento de las vasculitis fueron Rokitansky y Virchow en el siglo XIX, con la descripción de la poliarteritis nodosa. No obstante, generalmente se reconoce que fueron Kussmaul y Maier quienes en 1866 describieron por primera vez la enfermedad. Dichos autores llamaron la atención sobre un cuadro consistente en aneurismas arteriales nodulares, acompañados de un proceso inflamatorio en la adventicia, al que denominaron “periarteritis nodosa”. En 1903 Ferrari recuperó el término original de “poliarteritis nodosa” al observar el carácter multifocal y transmural del proceso.
Por otro lado, en 1910 Goodpasture describió a un paciente en el que demostró un cuadro de hemorragia pulmonar y glomerulonefritis con presencia de proliferación extracapilar (las así llamadas semilunas glomerulares). Su hallazgo permaneció en el olvido hasta 1958, cuando Stanton y Tange publicaron la descripción de un grupo de pacientes con características similares para los que acuñaron el término de síndrome de Goodpasture, que quedó así definido como hemorragia pulmonar asociada con nefritis. Sin embargo, la patogenia no quedó aclarada hasta 1964, cuando se encontró que algunos de esos pacientes presentaban un depósito lineal de inmunoglobulina (Ig) G que podía detectarse por inmunofluorescencia en la membrana basal del glomérulo renal. Desde entonces la presencia de hemorragias pulmonares se asoció en cierta manera con la enfermedad renal y con un tipo de afectación sistémica inflamatoria de los vasos sanguíneos, las llamadas vasculitis.
En 1937 Wegener descubrió en varios pacientes otro tipo de síndrome pulmonar renal, consistente en una vasculitis granulomatosa de las vías aéreas superiores y parénquima pulmonar, además de glomerulonefritis necrosante con semilunas. Fueron Godman y Churg quienes posteriormente bautizaron este síndrome como granulomatosis de Wegener. La enfermedad compartía características con la poliarteritis nodosa, pero era diferente porque tenía granulomas, los órganos que se afectaban no eran los mismos y se presentaba ocasionalmente en forma de hemorragia alveolar.
En los años setenta del siglo pasado, la combinación de hemorragia pulmonar y glomerulonefritis con semilunas, sin otros signos de arteritis, se agrupaba como una entidad única y concreta, mediada por anticuerpos antimembrana basal glomerular, y el resto de cuadros de hemorragia pulmonar con nefritis y arteritis, sin anticuerpos antimembrana basal glomerular, se clasificaban como granulomatosis de Wegener o poliarteritis nodosa, en una clasificación excesivamente simplista. La situación se complicó, ya que fueron describiéndose casos de hemorragia pulmonar y nefritis sin depósito de anticuerpos antimembrana basal glomerular, pero asociados con el depósito de inmunocomplejos (lupus eritematoso sistémico, crioglobulinemia mixta y púrpura de Henoch-Schonlein), con lo que se ampliaba el espectro de enfermedades y de mecanismos fisiopatológicos que podían producir inflamación vascular y/o hemorragia pulmonar. Actualmente para las vasculitis sistémicas se utiliza la clasificación de consenso propuesta en Chapel-Hill en 19942 (figs. 1 y 2).
")
Clasificación de las vasculitis. ANCA: anticuerpos citoplásmicos antineutrófilos; IF: inmunofluorescencia. (Tomada de Jenette et al2.)
. ANCA: anticuerpos citoplásmicos antineutrófilos; IF: inmunofluorescencia; IgA: inmunoglobulina A. (Tomada de Jenette et al2.)")
En patología pulmonar el cuadro clínico-patológico de la hemorragia alveolar difusa es extremadamente preocupante debido a su gravedad y a su heterogeneidad. Existen múltiples causas de hemorragia pulmonar, no sólo asociadas a vasculitis, y se ha hecho necesaria una sistematización centrada primordialmente en la utilidad diagnóstica y terapéutica.
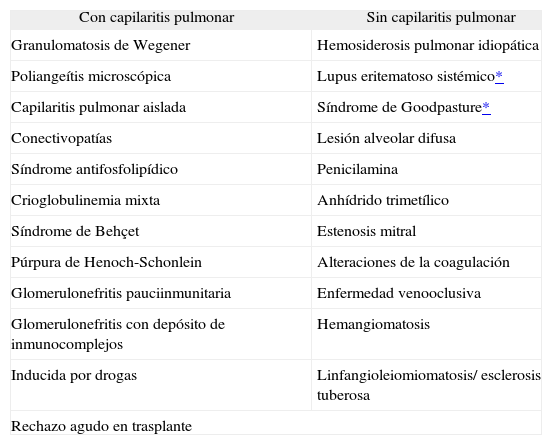
Causas de hemorragia alveolar difusaDesde el punto de vista del diagnóstico y del tratamiento puede resultar de utilidad dividir en 2 grandes grupos las hemorragias alveolares: las asociadas con fenómenos inflamatorios de la pequeña red capilar pulmonar (capilaritis), y aquéllas no asociadas a capilaritis3,4 (tabla I).
Hemorragias alveolares asociadas a capilaritis pulmonar y no asociadas a capilaritis
| Con capilaritis pulmonar | Sin capilaritis pulmonar |
| Granulomatosis de Wegener | Hemosiderosis pulmonar idiopática |
| Poliangeítis microscópica | Lupus eritematoso sistémico* |
| Capilaritis pulmonar aislada | Síndrome de Goodpasture* |
| Conectivopatías | Lesión alveolar difusa |
| Síndrome antifosfolipídico | Penicilamina |
| Crioglobulinemia mixta | Anhídrido trimetílico |
| Síndrome de Behçet | Estenosis mitral |
| Púrpura de Henoch-Schonlein | Alteraciones de la coagulación |
| Glomerulonefritis pauciinmunitaria | Enfermedad venooclusiva |
| Glomerulonefritis con depósito de inmunocomplejos | Hemangiomatosis |
| Inducida por drogas | Linfangioleiomiomatosis/ esclerosis tuberosa |
| Rechazo agudo en trasplante | |
Por lo tanto, si se toma como elemento guía para el diagnóstico diferencial una imagen morfológica, es preciso cuidar el aspecto que el tejido presentará en el examen microscópico. Este dato es de extrema importancia porque, si el paciente ha sido parcialmente tratado antes de realizar la biopsia, puede haber vasculitis que desde el punto de vista morfológico aparezcan como hemorragias alveolares sin capilaritis. Por este motivo hay que intentar tomar las muestras antes de que el paciente reciba ningún tratamiento, aunque, dadas las características graves de la enfermedad y la urgencia de la situación clínica debe informarse al anatomopatólogo de todo tipo de tratamiento administrado al paciente antes de la práctica de la biopsia.
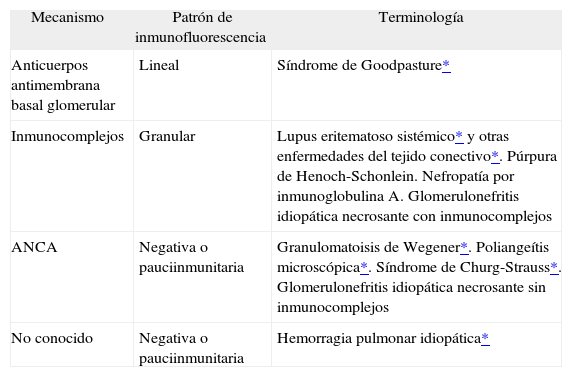
Dentro de cada uno de los 2 grupos mencionados (hemorragias asociadas y no a capilaritis), las causas de las hemorragias alveolares son muy variadas, pero es posible analizar varios datos para intentar alcanzar un diagnóstico5. La conjunción de la historia clínica -fundamental en todo tipo de enfermedad, pero mucho más en estos pacientes-, las determinaciones de laboratorio -anticuerpos citoplásmicos antineutrófilos (ANCA) y otros marcadoresy– y los datos que pueden obtenerse de la biopsia, tanto morfológicos como de inmunofluorescencia6 (tabla II), es lo que puede permitir un enfoque diagnóstico útil.
Datos morfológicos y de inmunofluorescencia que ayudan en el diagnóstico
| Mecanismo | Patrón de inmunofluorescencia | Terminología |
| Anticuerpos antimembrana basal glomerular | Lineal | Síndrome de Goodpasture* |
| Inmunocomplejos | Granular | Lupus eritematoso sistémico* y otras enfermedades del tejido conectivo*. Púrpura de Henoch-Schonlein. Nefropatía por inmunoglobulina A. Glomerulonefritis idiopática necrosante con inmunocomplejos |
| ANCA | Negativa o pauciinmunitaria | Granulomatoisis de Wegener*. Poliangeítis microscópica*. Síndrome de Churg-Strauss*. Glomerulonefritis idiopática necrosante sin inmunocomplejos |
| No conocido | Negativa o pauciinmunitaria | Hemorragia pulmonar idiopática* |
ANCA: anticuerpos citoplásmicos antineutrófilos.
Los ANCA, descritos en 1982 por Davis, tienen especificidad frente a proteínas que se encuentran en los gránulos de los polimorfonucleares y en los lisosomas peroxidasa positivos de los monocitos. Son marcadores, con una buena especificidad, de enfermedades como la granulomatosis de Wegener, la poliangeítis microscópica y la granulomatosis de Churg-Strauss. Se describen varias formas de ANCA, aunque son 2 las más importantes desde el punto de vista diagnóstico: la antiproteinasa-3 y la antimieloperoxidasa.
Existen 2 métodos de detección que se utilizan en la clínica: la inmunofluorescencia indirecta y los enzimoinmunoanálisis específicos. En la inmunofluorescencia indirecta se centrifugan polimorfonucleares en portas, se fijan en etanol y se incuban con el suero del paciente. Tras un lavado, se incuban con un anticuerpo secundario marcado con fluorescencia antiinmunoglobulina humana. En el caso de resultar positiva la prueba, se reconocen 3 patrones de inmunofluorescencia:
- 1.
Los ANCA con patrón citoplásmico (c-ANCA), que se caracterizan por mostrar una tinción citoplásmica granular que se acentúa entre los lóbulos y respeta el núcleo. En 1989 el antígeno reconocido por c-ANCA se identificó como una serinproteinasa sérica de 29kDa (proteinasa neutrofílica-3), presente en los gránulos azurófilos de los polinucleares y de los monocitos. La correlación del patrón c-ANCA con los anticuerpos frente a la proteinasa-3 es buena si interpretan la inmunofluorescencia personas con experiencia.
- 2.
Los ANCA con patrón perinuclear (p-ANCA), que tiñen el núcleo o el área perinuclear. Este patrón es un artefacto de redistribución de los antígenos citoplásmicos al núcleo durante la fijación con etanol, ya que los gránulos se rompen y las proteínas básicas cargadas positivamente migran hacia el núcleo, que muestra una carga electrostática negativa. Fueron Falk y Jenette quienes en 1988 describieron este segundo tipo de ANCA, que reconoce la mieloperoxidasa. Sin embargo, la correlación de los p-ANCA con la mieloperoxidasa no es del todo buena, ni siquiera cuando se tiene experiencia en la valoración de la tinción.
- 3.
Por último, existe un patrón atípico de ANCA (en tormenta de nieve), que no se asocia con ningún antígeno específico, aunque en algunos pacientes se aprecia reacción frente a lactoferrina, lisozima, betaglucuronidasa o catepsina G.
La mayor frecuencia de positividad para ANCA se da en los pacientes que no están en tratamiento y que presentan enfermedad activa. Tanto el título como la frecuencia de positividad disminuyen al instaurar el tratamiento inmunodepresor y entrar la enfermedad en una fase quiescente.
Biopsia pulmonar e inmunofluorescenciaLa misión fundamental de la biopsia pulmonar en los casos de sospecha de hemorragia pulmonar es triple. En primer lugar, mediante métodos morfológicos debe confirmar la hemorragia alveolar y descartar un cuadro inflamatorio de vaso pequeño (capilaritis). En segundo lugar, debe descartar la presencia de otras causas no vasculíticas de hemorragia. En tercer lugar, debe proporcionar el dato de los posibles depósitos inmunitarios, detectados mediante inmunofluorescencia7. Es muy discutible el posible aumento de la morbimortalidad quirúrgica en estos pacientes debido a la toma de biopsia, ya que no hay evidencias que indiquen dicho incremento4.
El estudio de inmunofluorescencia en tejido pulmonar requiere disponer de tejido en fresco, ya que, aunque hay anticuerpos marcados con peroxidasa frente a inmunoglobulinas que pueden aplicarse sobre tejido fijado en formol e incluido en parafina, generalmente no proporcionan un buen resultado. Por tanto, es necesario congelar un fragmento de parénquima pulmonar para realizar la técnica de inmunofluorescencia frente a IgA, IgG, IgM, las fracciones de complemento C3, C4 y C1q y frente a fibrinógeno y albúmina. Es preciso destacar que la valoración de estas técnicas es mucho más complicada que en el tejido renal, debido al elevado contenido en fibras elásticas del pulmón, a la autofluorescencia de éste y a la presencia de otras lesiones como hemorragia, inflamación o exudados que pueden producir errores de interpretación.
Es la integración de toda la información obtenida del estudio serológico con la morfología e inmunofluorescencia la que permitirá dividir los cuadros en asociados a capilaritis o no, y en pauciinmunitarios (sin o escasos depósitos inmunológicos) o asociados a depósitos inmunitarios.
Últimamente se discute de nuevo el valor de la biopsia transbronquial para el diagnóstico de la enfermedad pulmonar intersticial difusa. Si bien es cierto que la biopsia por videotoracoscopia es la mejor solución porque permite un estudio morfológico e inmunológico completo, la biopsia transbronquial puede resultar de utilidad y proporcionar datos que favorezcan o descarten determinadas enfermedades estrechando el diagnóstico diferencial y ofreciendo opciones de tratamiento8,9 en determinados casos.
Cuadros pauciinmunitariosEn este apartado se incluyen los síndromes asociados con ANCA: la granulomatosis de Wegener, la poliangeítis microscópica, el síndrome de Churg-Strauss, la glomerulonefritis necrosante con semilunas y con/sin hemorragia pulmonar y las formas mixtas10. Ocurren más comúnmente en varones de 50 a 60 años. Morfológicamente se caracterizan por mostrar capilaritis o vasculitis de pequeño vaso. Sin embargo, la presencia de capilaritis en una biopsia pulmonar no es del todo específica, y este fenómeno puede observarse en pacientes con síndromes vasculíticos sin hemorragia alveolar11. Las 2 características que indican más fehacientemente la capilaritis son la acumulación de polimorfonucleares en el intersticio, más allá de la proporción que le correspondería por la presencia de las mismas células en el espacio alveolar, así como la existencia de restos celulares (cariorrexis) de los mismos12 (fig. 3). Por definición, la inmunofluorescencia no debe demostrar depósitos inmunológicos en capilares pulmonares.
")
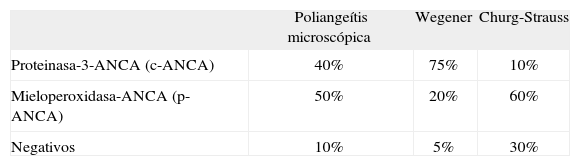
La capilaritis es un reflejo de daño vascular y generalmente los ANCA están implicados en su patogenia. Sin embargo, como en cualquier prueba diagnóstica, la sensibilidad no alcanza el 100%, ya que no todos los casos de las enfermedades aquí descritas muestran positividad frente a ANCA (tabla III). Por otro lado, la especificidad tampoco es del 100%, pues hay infecciones hematógenas, particularmente en pacientes inmunodeprimidos, que en ocasiones muestran ANCA en sangre periférica, al igual que ocurre en pacientes con enfermedad inflamatoria intestinal y otras enfermedades autoinmunitarias como el lupus eritematoso sistémico y la artritis reumatoide11. Por tanto, no debe utilizarse de manera aislada una determinación positiva para ANCA como diagnóstica de granulomatosis de Wegener o poliangeítis microscópica ante un paciente que no cumple claramente los criterios establecidos13.
Positividad de anticuerpos en los cuadros pauciinmunitarios
| Poliangeítis microscópica | Wegener | Churg-Strauss | |
| Proteinasa-3-ANCA (c-ANCA) | 40% | 75% | 10% |
| Mieloperoxidasa-ANCA (p-ANCA) | 50% | 20% | 60% |
| Negativos | 10% | 5% | 30% |
ANCA: anticuerpos citoplásmicos antineutrófilos (c-ANCA: con patrón citoplásmico; p-ANCA: con patrón perinuclear).
La misión de esta revisión no es mostrar en detalle todos los aspectos morfológicos de estas enfermedades, sino la de poner de manifiesto que todas ellas pueden tener como signo de inicio una hemorragia alveolar difusa.
Granulomatosis de WegenerSe define como una inflamación granulomatosa que afecta al aparato respiratorio y una vasculitis que afecta a vasos sanguíneos de tamaño mediano y pequeño. La forma de presentación clásica es bien conocida. Sin embargo, la hemorragia alveolar difusa secundaria a vasculitis de pequeño vaso puede ser la manifestación inicial y única de la granulomatosis de Wegener14. Se han comunicado aproximadamente unos 40 casos con estas características4. Estos pacientes suelen presentarse con afectación renal grave, de modo que la asociación de dicha insuficiencia renal puede resultar de ayuda diagnóstica. La mortalidad temprana es considerablemente mayor en los pacientes cuyo signo de inicio es una hemorragia alveolar, tanto por la gravedad de la situación como por las infecciones secundarias al tratamiento inmunodepresor agresivo que se aplica.
Poliangeítis microscópicaSegún la clasificación de Chapel-Hill, es una vasculitis necrosante con pocos o ningún depósito inmunitario, que afecta a pequeños vasos. Esta definición incluye casos que en clasificaciones anteriores figuraban como vasculitis por hipersensibilidad o poliarteritis nodosa. La lesión pulmonar más común es la capilaritis con hemorragia alveolar, con o sin arteritis necrosante (fig. 4).
")
Las características diferenciales con la poliarteritis nodosa clásica son, además del calibre del vaso afectado, que la poliangeítis microscópica casi nunca se asocia con una infección previa por hepatitis B o C; que la afectación pulmonar es infrecuente en la variante clásica, mientras que existe en un 10-30% de los pacientes con la forma microscópica, y que la glomerulonefritis rápidamente progresiva, que se da en un 80-100% de los casos de poliangeítis microscópica, no ocurre en la poliarteritis nodosa.
La afectación pulmonar incrementa la mortalidad temprana de la poliangeítis microscópica. De hecho, hasta un 25% de los pacientes fallecen durante el primer episodio de hemorragia alveolar4. Entre los que sobreviven al primer episodio, ante los episodios recidivantes de hemorragia, desarrollan enfermedad obstructiva (se piensa que enfisema, aunque no está bien demostrado) y fibrosis pulmonar. Por otro lado, en los lugares donde ha habido inflamación vascular necrosante se desarrolla una esclerosis vascular con disrupción de las fibras elásticas.
Síndrome de Churg-StraussSe define como una inflamación granulomatosa rica en eosinófilos que afecta al aparato respiratorio, junto con una vasculitis necrosante que afecta a vasos de mediano y pequeño calibre, asociada con asma y eosinofilia. La situación habitual en el diagnóstico es que la biopsia sea indicativa o no de un diagnóstico establecido basado en criterios clínicos. Por tanto, el anatomopatólogo no confirma o excluye un diagnóstico de síndrome de Churg-Strauss, sino que orienta basándose en criterios morfológicos15. La presencia de hemorragia alveolar es mucho menos común que en el resto de vasculitis de vaso pequeño11.
Capilaritis pulmonar aisladaEs un síndrome de hemorragia alveolar difusa que puede aparecer en pacientes con o sin positividad para p-ANCA. Los escasos pacientes descritos con ANCA negativos no desarrollaron enfermedad sistémica clínica ni serológica tras un seguimiento prolongado4,16. Todos los pacientes mostraron enfermedad limitada a los pulmones. Se ha planteado la posibilidad de que sea un equivalente pulmonar de la glomerulonefritis pauciinmunitaria idiopática. Estos casos son difícilmente distinguibles de la poliangeítis microscópica, y su existencia y naturaleza continúan siendo una cuestión controvertida.
Cuadros asociados a depósitos inmunitariosEnfermedad por anticuerpos antimembrana basal glomerular (sin anticuerpos citoplásmicos antineutrófilos)La diana frente a la que se dirigen los anticuerpos antimembrana basal glomerular son epítopos en el dominio NC1 de la cadena α3 del colágeno tipo IV (antígeno de Goodpasture).
El diagnóstico característico se realiza con biopsia pulmonar e inmunofluorescencia, al detectar un patrón lineal de IgG en la membrana basal capilar del pulmón, semejante al que se obtiene en la biopsia renal, donde se observa un depósito lineal de IgG en el glomérulo renal. Sin embargo, este hallazgo no es del todo específico porque la IgG puede acumularse de forma lineal de una manera no específica. La clave en estos casos la da la tinción con albúmina, que si proporciona un patrón similar al de IgG correspondería a un patrón no característico de la enfermedad por anticuerpos antimembrana basal glomerular.
En algunos casos aislados puede haber también positividad para ANCA. La enfermedad es muy poco frecuente (0,5 casos por millón/año) y suele afectar a pacientes varones de alrededor de 40 años, generalmente fumadores.
Morfológicamente se manifiesta como un cuadro de hemorragia alveolar sin arteritis, aunque puede haber capilaritis focal. La trama alveolar de fibras reticulínicas y elásticas está preservada. El depósito de IgG en la membrana basal alveolar del pulmón puede demostrarse a veces por inmunofluorescencia, pero suele ser de escasa entidad17.
No todos los pacientes con enfermedad por anticuerpos antimembrana basal glomerular desarrollan lesiones pulmonares, de modo que parece que podría existir algún otro factor subyacente independiente de los anticuerpos, como el tabaco o la presencia de citocinas inflamatorias18.
Enfermedades asociadas con otros depósitos inmunitariosLas enfermedades del colágeno son otra causa relativamente frecuente de hemorragias pulmonares19. Estas enfermedades son de diagnóstico difícil debido a su presentación inespecífica. Muchas veces son precisas múltiples pruebas de imagen, serologías frente a diferentes anticuerpos y los hallazgos morfológicos de la biopsia, por lo que es preciso conocer la variedad de enfermedades de este tipo que pueden presentarse como hemorragias alveolares para facilitar un rápido diagnóstico diferencial.
La hemorragia alveolar difusa es una complicación de los pacientes con lupus eritematoso sistémico en un 4% de los casos. Llega a ser el síntoma de presentación de la enfermedad hasta en un 23% y es la causa más frecuente de mortalidad, que alcanza un 50% de los casos en que se presentó este fenómeno. Cuando la hemorragia alveolar se manifiesta una vez diagnosticado el lupus, lo hace una media de 30 meses después. Es llamativo el hecho de que en sólo el 42-66% de los casos se describe hemoptisis como síntoma inicial. Sin embargo, sí aparece casi de forma constante la nefritis lúpica como signo acompañante20. El diagnóstico diferencial fundamental es con un cuadro infeccioso, que de hecho es la causa más frecuente de enfermedad parenquimatosa pulmonar en estos pacientes. témica semejante a la poliangeítis microscópica. El mecanismo de la trombosis incluye la presencia de anticuerpos dirigidos frente a antígenos de células endoteliales, inmunocomplejos circulantes depositados en el endotelio y otros factores que afectan a la coagulación como el anticoagulante lúpico21.
Los depósitos de IgG y de fracciones del complemento en las paredes alveolares son visibles de una manera granular (fig. 5). En el caso de que los depósitos sean únicamente de IgA, se debería considerar un diagnóstico de hemorragia alveolar en el seno de una púrpura de Henoch-Schonlein o de una nefropatía por IgA4,22. Esta asociación ocurre en menos del 5% de los casos2, pero esto quizá sea debido a que no se ha estudiado un gran número de casos con biopsia e inmunofluorescencia, ya que, como en cualquier enfermedad por inmunocomplejos, éstos son circulantes y, por tanto, susceptibles de depositarse en el pulmón, como ya se ha demostrado en casos diagnosticados de otro tipo de enfermedad intersticial como las bronquiolitis23.
 A, B y C: imágenes de inmunofluorescencia que evidencian doble depósito granular en el glomérulo renal (B: anticuerpo antiinmunoglobulina G; C: anti-C3. Aumentos originales: ×100). D: depósito granular en capilares pulmonares (anti-C3. Aumentos originales: ×340).")
Hemorragia alveolar sin capilaritis en un caso de lupus eritematoso sistémico. (HE, ×10.) A, B y C: imágenes de inmunofluorescencia que evidencian doble depósito granular en el glomérulo renal (B: anticuerpo antiinmunoglobulina G; C: anti-C3. Aumentos originales: ×100). D: depósito granular en capilares pulmonares (anti-C3. Aumentos originales: ×340).
La artritis reumatoide es una enfermedad inflamatoria crónica con manifestaciones en tejidos extraarticulares que incluyen el pulmón. En los pacientes con afectación extraarticular el factor reumatoide suele ser positivo. La vasculitis es infrecuente (< 10% de los pacientes)10 y puede afectar a todo tipo de vasos sanguíneos con fenómenos trombóticos. El infiltrado inflamatorio es de densidad variable y puede ser linfoide, granulomatoso o neutrofílico, y acompañarse de hemorragias pulmonares24.
También se han descrito cuadros de hemorragia alveolar, aunque extremadamente infrecuentes, en pacientes con polimiositis, enfermedad mixta del tejido conectivo y esclerodermia25,26.
Dentro de los cuadros más graves que pueden producir hemorragia alveolar y que se asocian a enfermedad inmunológica se encuentra la variante catastrófica del síndrome antifosfolipídíco (fig. 6). Afecta predominantemente a mujeres (66%), no en todos los casos hay un síndrome antifosfolipídico clásico asociado y puede aparecer en relación con cuadros de lupus eritematoso sistémico, síndrome seudolúpico o artritis reumatoide. Existe una forma de presentación pediátrica y se ha descrito una serie de factores precipitantes asociados (infecciones, drogas, cirugía menor, etc.). Clínicamente se aprecia microangiopatía con insuficiencia multiorgánica, trombocitopenia, anemia hemolítica, coagulación intravascular diseminada y presencia de esquistocitos en sangre periférica.
")
En las exploraciones de laboratorio pueden aparecer anticoagulante lúpico, anticuerpos anticardiolipina, anti-ADN de doble cadena, anticuerpos antinucleares, anti-Ro/ SS-A, anticuerpos antirribonucleoproteína y anti-LA/SS-B. Se manifiesta en forma de fenómenos trombóticos en todo el árbol vascular. Es extremadamente grave, con un 50% de mortalidad a pesar del tratamiento con anticoagulación, esteroides, plasmaféresis o gammaglobulinas intravenosas27,28.
Otras causas de hemorragia pulmonar. MisceláneaExisten otras causas de hemorragia alveolar difusa, tales como la lesión alveolar difusa, la enfermedad venooclusiva, la hemorragia pulmonar no inflamatoria que puede ocurrir en la estenosis mitral, como complicación de anticoagulación y tratamiento trombolítico29,30 (en estos 2 últimos casos con histología normal), uso de ácido transretinoico en la leucemia promielocítica31, coagulopatías, toxicidad por otros fármacos como el gemtuzumab32, infliximab33, sirolimus34, everolimus, penicilamina35, rituximab36, vasculitis inducidas por agentes infecciosos, complicaciones no infecciosas tras un trasplante de médula ósea37 y hemosiderosis pulmonar idiopática.
Es precisa una historia clínica exhaustiva que incluya el consumo de drogas como el crack (cocaína)38, que produce hemorragia por lesión alveolar difusa, y el propiltiouracilo o la difenilhidantoína, que causan vasculitis por hipersensibilidad39.
Los casos atribuidos a toxicidad por el uso de cocaína se asocian a un cuadro exuberante de lesión alveolar difusa, con fenómenos organizativos y un componente importante de macrófagos alveolares cargados de pigmento férrico más grueso que el que se ve habitualmente en los casos de hemorragia por otras causas. En caso de sospecha, la biopsia transbronquial puede ser extremadamente útil al demostrar un patrón morfológico de hemorragia sin capilaritis.
En cuanto a la toxicidad por fármacos como la penicilamina, además de la hemorragia alveolar suelen aparecer otros patrones de lesión habituales en los casos toxicidad, como la bronquiolitis, la hiperplasia linfoide o áreas de neumonía organizativa, casi siempre sin vasculitis. Ha sido controvertido el papel de la penicilamina en el desarrollo de lesiones pulmonares, ya que la mayor parte de los casos se han descrito en pacientes tratados por artritis reumatoide y en ocasiones las lesiones que producen tanto el fármaco como la propia enfermedad son similares. Sin embargo, otros pacientes con el mismo tipo de tratamiento, como los afectados por la enfermedad de Wilson, muestran lesiones de hemorragia, bronquiolitis obliterante e hiperplasia linfoide, con lo que el papel del fármaco en la producción de toxicidad se pone de manifiesto.
Por otro lado, hay otras causas mucho más infrecuentes de hemorragia alveolar difusa, pero que deben tenerse presente, como la de los receptores de trasplantes de sangre de cordón umbilical para tratamiento de enfermedades raras, procedimiento que, al igual que el trasplante de células progenitoras, se utilizará de forma más habitual conforme avance el tiempo40.
En el seno del trasplante de células madre hematopoyéticas aparece asimismo de manera relativamente frecuente una situación de hemorragia alveolar difusa41. Se caracteriza por un cuadro de aparición brusca consistente en disnea, tos e hipoxemia, con o sin fiebre. La hemoptisis franca es infrecuente. La hemorragia puede asociarse a 3 situaciones. En primer lugar, puede aparecer un cuadro de hemorragia alveolar en el período temprano después de un trasplante generalmente autólogo. Su incidencia es de un 7-14% y parece que se asocia con la presencia de enfermedad del injerto contra el huésped. Esta situación se ha asociado con una mortalidad del 75% a pesar de tratamiento. En segundo lugar, y de nuevo en el período temprano, tras un trasplante autólogo puede desarrollarse un síndrome de distrés respiratorio con características de lesión alveolar difusa y hemorragia, que responde bien al tratamiento esteroideo. La tercera situación asociada a hemorragia alveolar es la del síndrome de toxicidad pulmonar retardada, que ocurre meses después del trasplante, tiene escasa respuesta a tratamiento42 y podría estar relacionado con el acondicionamiento previo al trasplante.
La aparición de hemorragias pulmonares secundarias a vasculitis sépticas es una complicación conocida de los agentes bacterianos, víricos y fúngicos. De hecho, las neumonías hemorrágicas en pacientes inmunodeprimidos tienen frecuentemente una etiología infecciosa, y entre los agentes más frecuentes se encuentran los hongos del tipo Aspergillus en su forma angioinvasiva43 y una forma especial fúngica conocida como Scedosporium prolificans, importante en pacientes hematológicos44. Entre los agentes bacterianos, quizá la más conocida sea la causada por leptospirosis45. Por su parte, agentes víricos como el citomegalovirus o los virus de la hepatitis B y C pueden producir complicaciones hemorrágicas, tanto por acción patogénica directa (citomegalovirus) como por mecanismos indirectos (crioglobulinemia mixta y hepatitis B y C)46,47.
A este respecto parece necesaria una reflexión, pues la mayor parte de las vasculitis en humanos entendemos que son idiopáticas o autoinmunitarias, en contraste con el mundo animal, donde los agentes infecciosos causantes de vasculitis son bien conocidos. Probablemente la utilización de técnicas moleculares para la detección de patógenos, tanto víricos como bacterianos, en el propio tejido lesionado proporcione datos interesantes para la patogenia y el tratamiento de estos pacientes48.
La última causa de hemorragia alveolar difusa es la hemosiderosis pulmonar idiopática. Es una enfermedad que suele presentarse en niños menores de 16 años y que a veces se asocia con celiaquía y un aumento de los títulos de IgA. Desde el punto de vista morfológico se manifiesta por ausencia de vasculitis y de afectación renal. La lesión clásica es una inflamación no específica, no granulomatosa, indistinguible del síndrome de Goodpasture morfológicamente, sin depósito de inmunocomplejos. Debido a la inespecificidad tanto clínica como morfológica, el diagnóstico es de exclusión49. Se han comunicado casos donde los corticoides e inmunodepresores pueden ser efectivos.
En resumen, las hemorragias alveolares difusas son cuadros graves que pueden ser catastróficos si no se diagnostican y tratan a tiempo. Las claves diagnósticas son una buena historia clínica y el conocimiento e integración de estudios serológicos y morfológicos.







