









Idiopathic pulmonary fibrosis is defined as a chronic fibrosing interstitial pneumonia limited to the lung, of unknown cause, with poor prognosis and few treatment options. In recent years there has been an increase in their prevalence, probably due to the optimization of diagnostic methods and increased life expectancy. The ATS/ERS Consensus (2000) established the diagnostic criteria and recommendations for the assessment of the disease course and treatment. Later studies have helped to redefine diagnostic criteria and treatment options. In 2011, an international consensus was published, establishing diagnostic criteria and new treatment strategies. These guidelines have been updated with the newest aspects of diagnosis and treatment of idiopathic pulmonary fibrosis. A level of evidence has been identified for the most relevant questions, particularly with regard to treatment options.
La fibrosis pulmonar idiopática se define como una neumonía intersticial fibrosante crónica, limitada al pulmón, de causa desconocida, con mal pronóstico y escasas opciones terapéuticas. En los últimos años se ha observado un incremento en su prevalencia, probablemente debido a la optimización de los métodos diagnósticos y al aumento de la esperanza de vida. En el consenso ATS/ERS del año 2000 se establecieron por primera vez los criterios diagnósticos y las recomendaciones para evaluar su evolución y tratamiento. Posteriormente, diversos estudios han contribuido a optimizar las pautas diagnósticas y terapéuticas. En el año 2011 se publicó un consenso internacional en el que se redefinieron los criterios diagnósticos y se establecieron nuevas recomendaciones terapéuticas. En esta normativa se actualizan los aspectos novedosos del diagnóstico y el tratamiento de la fibrosis pulmonar idiopática. Se ha atribuido un nivel de evidencia a las cuestiones más relevantes, principalmente en el apartado dedicado al tratamiento.
The ATS/ERS consensus published in 2000 on idiopathic pulmonary fibrosis (IPF) set out, for the first time, the diagnostic criteria and recommendations for evaluating its course and treatment.1 Since its publication, a number of studies have contributed to optimizing the diagnostic and therapeutic guidelines for IPF. As a result, an international consensus was published in 2011, in which the diagnostic criteria were redefined and new therapeutic recommendations were established.2 In 2003, the SEPAR Research group on Diffuse Pulmonary Diseases drew up guidelines on the Diagnosis and Treatment of Diffuse Interstitial Lung Diseases (DILD).3 In order to update these Guidelines, we thought it appropriate to limit them to IPF, as this is one of the diseases in which there have been most changes and developments in recent years. The present guidelines detail the changes that have occurred in the diagnostic criteria and in new therapeutic strategies. The GRADE system has been used to determine the recommendations and evidence.4
DefinitionIPF is defined as specific form of chronic, fibrosing interstitial pneumonia of unknown cause, typically affecting adults over 50 years old, limited to the lung, and associated with the histopathologic and/or radiologic pattern of usual interstitial pneumonia (UIP).2
Incidence and PrevalenceVarious studies have been conducted to evaluate the incidence and prevalence of IPF. The most reliable data estimate that the incidence varies between 4.6 and 7.4 cases/100000, and the prevalence is between 13 cases/100000 for females and 20 cases/100000 for males.2 Based on these data, it is believed that, in Spain, IPF could be affecting around 7500 people. It is unknown whether the incidence and prevalence are influenced by ethnic, racial or geographical factors. An increase in the prevalence has been observed in recent years, probably due to the optimization of diagnostic methods and an increase in life expectancy.5,6
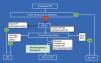
Natural HistoryThe natural history of IPF is variable and unpredictable at the time of diagnosis (Fig. 1). Some patients can remain asymptomatic for 2–3 years. However, most present a slow progression with clinical and functional deterioration that eventually results in chronic respiratory failure. In other cases, there are periods of relative stability with episodes of acute worsening (acute exacerbations or other complications), which are a cause of high morbidity and mortality.2 In a minority of the patients, the disease is of short duration with more rapid progression (accelerated form).7 In general, the average survival is 2–5 years from symptom onset.2 It is unknown whether the different forms of natural history represent different disease phenotypes.
Etiology and Risk Factors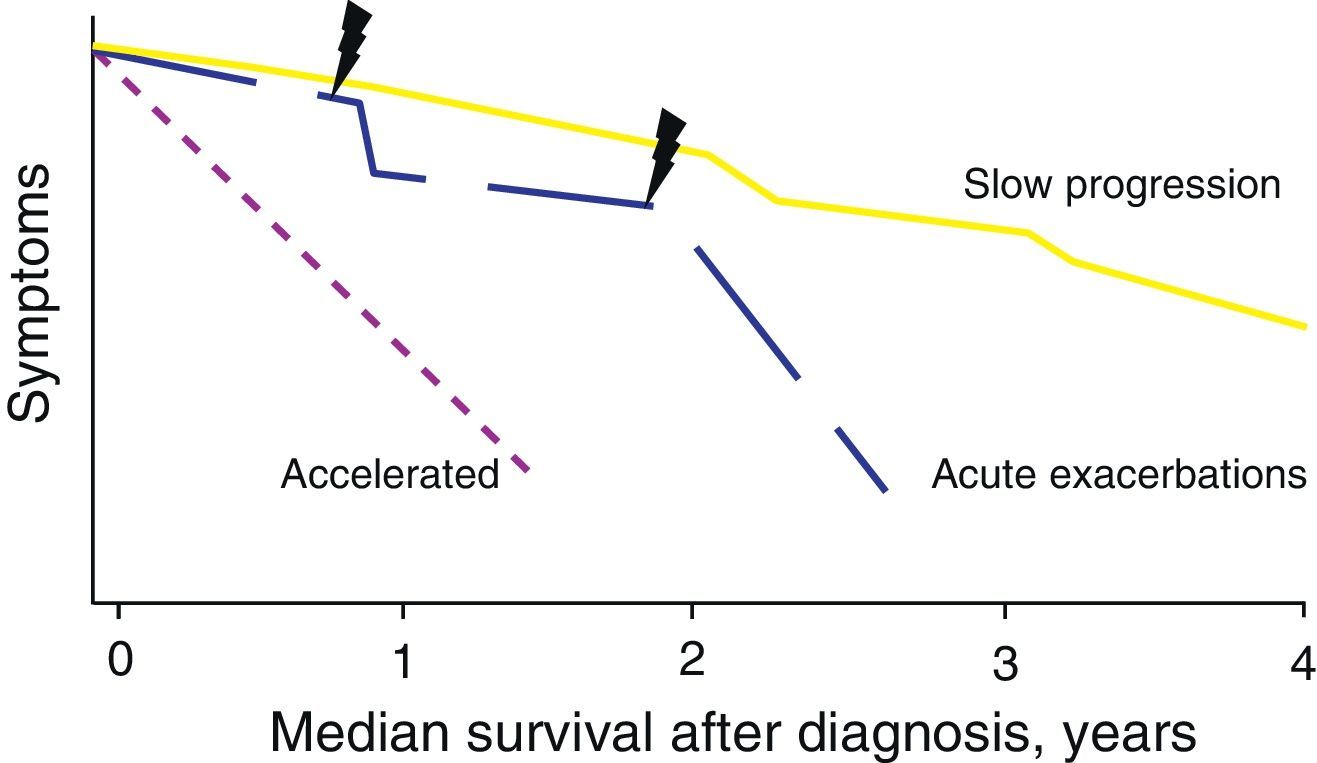
The etiology of IPF is unknown, although it is probably due to the effect of various factors in genetically predisposed subjects.8
Genetic FactorsThe most clinically relevant genetic alterations are: mutations in the genes that maintain the length of the telomeres (TERT, TERC), which are more common in the familial forms, in the surfactant protein C gene and in the mucin 5B promoter region (MUC5B). There are no established genetic tests to assess predisposition to IPF.8,9
Environmental FactorsSmoking (>20 packs/year) and exposure to silicon, brass, steel, lead and wood dust, farming and agricultural work, and the construction of wooden houses are considered risk factors.8,10,11
Gastroesophageal RefluxSeveral studies have shown that gastroesophageal reflux is a risk factor for the predisposition and progression of IPF.12
Viral InfectionsThere is insufficient evidence to consider that viral infections (hepatitis C virus, herpes virus, adenovirus) are etiological risk factors for IPF, although their contribution remains under study.
AutoimmunityThe possible autoimmune origin of IPF is based on the fact that the radiologic and/or histologic manifestations of UIP are associated with connective tissue diseases, although these usually present with histology typical of non-specific interstitial pneumonia.
DiagnosisThe definitive diagnosis of IPF requires: (a) the exclusion of other defined clinical entities or diffuse parenchymal lung diseases of known cause (environmental or occupational exposure, connective tissue diseases, drug toxicity) and (b) the presence of a histological pattern of UIP in the examination of lung tissue obtained by surgical lung biopsy, or radiological evidence of a UIP pattern on the high resolution computed tomography (HRCT), or both.
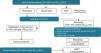
Multidisciplinary assessment in which pulmonologists, radiologists and pathologists experienced in the diagnosis and management of DILD can increase the diagnostic accuracy, which at present is a widely accepted recommendation for establishing the diagnosis (Fig. 2).2,13
Clinical Characteristics and Additional Tests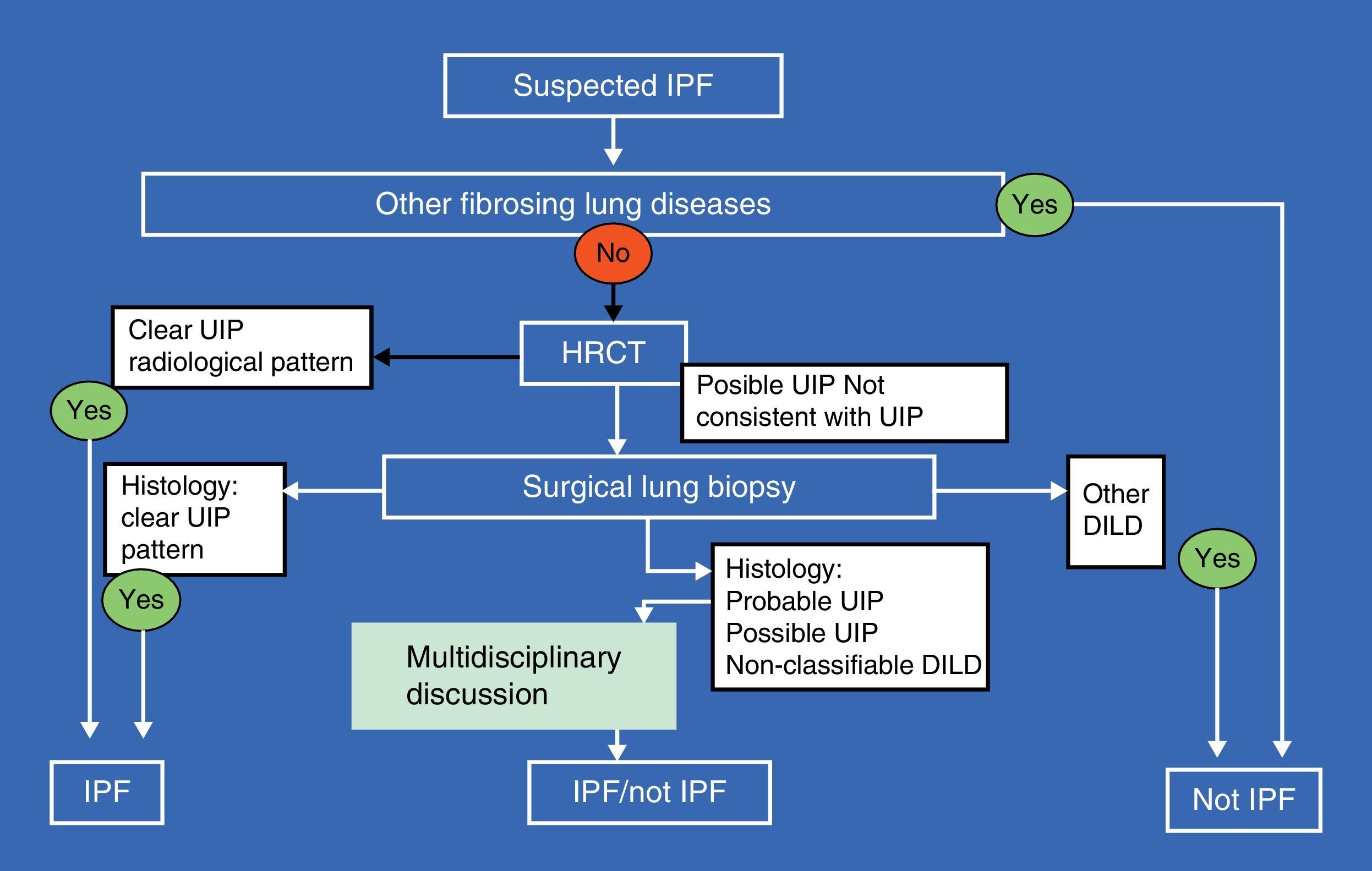
The clinical presentation of IPF has an insidious onset, and is usually characterized by progressive dyspnea on exertion, often accompanied by non-productive cough. Symptom onset is slow, but worsens over time. The delay between the onset of symptoms and the final diagnosis is variable, and may be between 6 months and two years.14 The presence of systemic symptoms/signs should lead to suspicion of an alternative diagnosis. Crackles can be heard on auscultation in 90% of patients and acropachy is detected in 50%.3
There are no specific laboratory abnormalities for this disease. However, even in the absence of specific signs or symptoms of connective tissue diseases, serological autoimmune tests should be performed in all patients.2 In fact, positive anti-nuclear antibodies or rheumatoid factor can be detected in up to 20% of IPF cases.3 The presence of serum specific IgG against the antigens that can most often cause hypersensitivity pneumonitis should be assessed systematically, since its clinical manifestations are occasionally similar to those of IPF. If any of these are positive, in the context of plausible exposure and bronchoalveolar lavage (BAL) with an increased lymphocyte count, a provocation test against the antigen in question and/or surgical lung biopsy should be performed, in order to confirm or discard the diagnosis of chronic hypersensitivity pneumonia.3 The possibility of using new biomarkers in the diagnosis and characterization of this disease has also gained interest in recent years. Some such as KL-6, SP-A and SP-D, circulating fibrocytes and metalloproteinases 1 and 7 are currently being researched.15–18
Bronchoalveolar Lavage and Transbronchial BiopsyBronchoalveolar lavage (BAL) has been extensively used in the study of DILD. Its analysis in IPF usually shows discrete neutrophilia with or without eosinophilia, and is use has been classically related with its ability to rule out other entities.2,3 The last international consensus indicated that BAL cellular analysis should not be performed systematically in all patients in the diagnostic procedure, although it may be appropriate for a minority.2 Nevertheless, in certain cases, BAL may be very helpful in the differential diagnosis with other entities such as chronic hypersensitivity pneumonitis or non-specific interstitial pneumonia.
Transbronchial biopsy is useful in diseases with lymphatic and centrilobular distribution, or in those that present characteristic diagnostic features and which have a diffuse distribution.19 It is being increasingly used for the diagnosis of sarcoidosis, infections and tumors.19 In contrast, it is of no use in the diagnosis of IPF, since the distribution of the lesion cannot be observed due to the sample size. The incorporation of cryobiopsy to the procedure is very promising, but further studies are required to corroborate its usefulness in DILD.20
High Resolution Computed Axial TomographyHRCT represents possibly the greatest diagnostic advance of the last two decades in the study of diffuse lung diseases. HRCT, either by sequential (slice-by-slice acquisition) or volumetric acquisition (continuous acquisition) is the undisputed technique in the diagnosis of IPF. Knowing the radiation dose used in HRCT is very important; the radiation dose used in volumetric HRCT is triple the values reached using sequential HRCT. The decision to use one or other technique will depend on the balance between the information expected and the individual risk due to the increased radiation received. Following established protocols is recommended, taking into account that the patient's age and sex are decisive factors (e.g. sequential HRCT, with 10mm intervals, in the initial assessment of patients under 40 years, and multi-detector computed tomography (MDCT) in patients aged 50 years or over).21
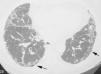
The objective is to identify findings typical of the UIP pattern (Fig. 3), and to distinguish them from the less specific patterns present in other idiopathic interstitial pneumonias. To prevent descriptive and conceptual errors, the radiological reading should use descriptive terminology based on the radiological–pathological correlation, as recommended by the Fleischner Society.22
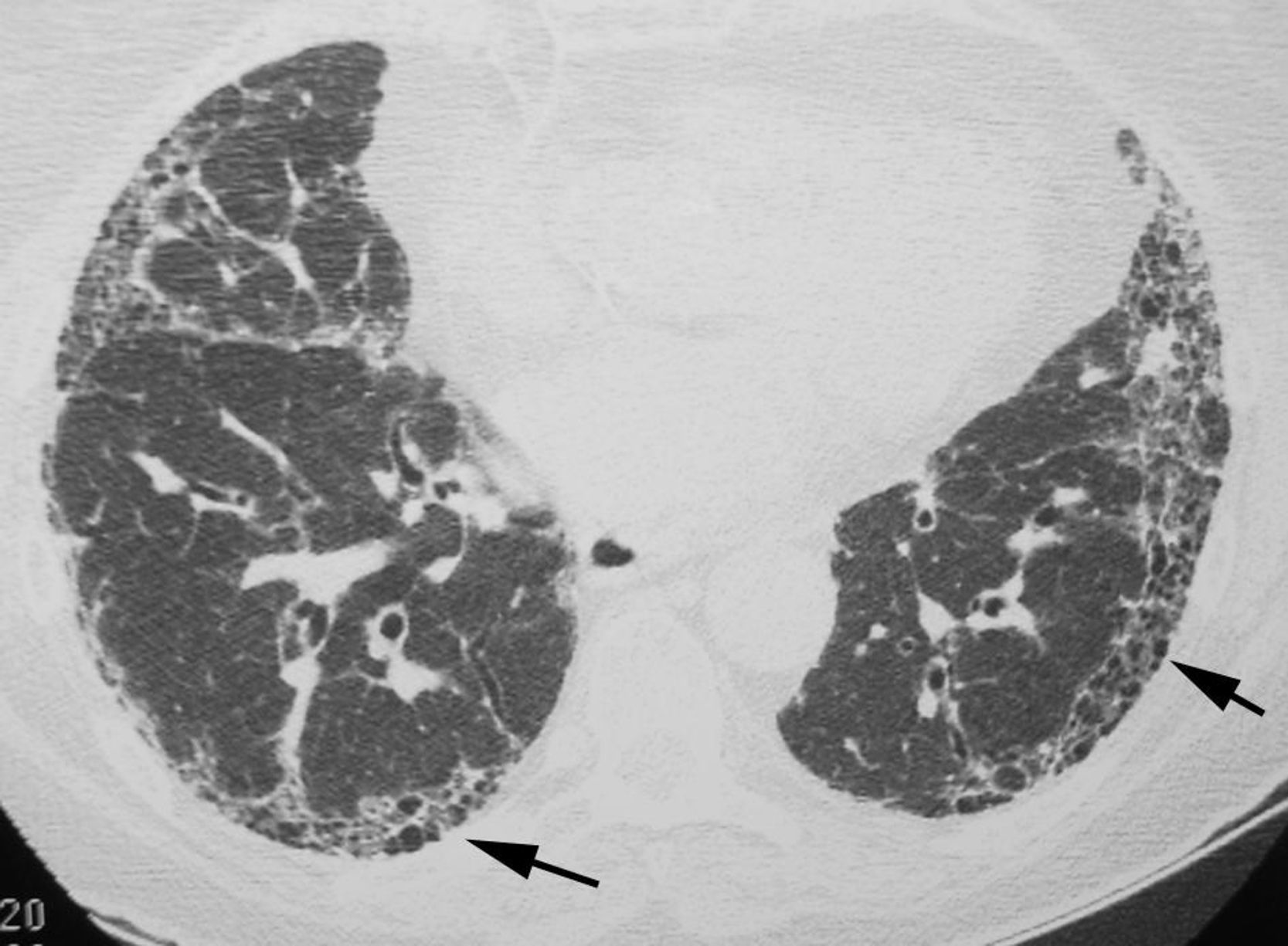
The official 2011 ATS/ERS/JRS/ALAT consensus2 established that in HRCT, the definite diagnosis of UIP is based on the identification of four “typical” findings: (1) lung involvement should have subpleural and basal predominance, (2) presence of obvious reticulation, (3) existence of honeycombing with/without traction bronchiectasis/bronchiolectasis and (4) demonstrate the absence of findings considered to exclude a UIP pattern (Table 1, Fig. 3). The presence of ground glass opacities should be minimal or non-existent.
HRCT Findings Considered to Exclude a UIP Pattern.
| Predominance in middle or upper fields |
| Peri-bronchovascular predominance |
| Significant presence of ground glass opacities |
| Numerous micronodules (bilateral, upper lobes) |
| Cysts (multiple, bilateral, distant from the areas of honeycombing) |
| Mosaic attenuation pattern/air trapping (bilateral, in three or more lobes) |
| Segment consolidation |
Adapted from Raghu et al.2
Honeycombing, made up of groups of thin-walled cysts, subpleural with a diameter between 3 and 10mm, is an essential finding for accurately diagnosing the UIP pattern. When there is no visible honeycombing, the diagnosis by HRCT will be that of a possible UIP pattern; in these cases, the definitive diagnosis of UIP should be made by biopsy. Lung biopsy can be avoided only when the HRCT shows a definitive pattern, typical of UIP. The positive predictive value of HRCT in the diagnosis of UIP is 90%–100%. A UIP pattern can also be identified in chronic hypersensitivity pneumonitis, asbestosis and some connective tissue diseases.23
HRCT also enables the presence of associated comorbidities (emphysema, pulmonary hypertension, lung cancer), which may determine the clinical progression of the disease, to be assessed. Other diffuse lung diseases, apart from idiopathic interstitial pneumonias, can also be suspected by HRCT.
The 2011 official ATS/ERS/JRS/ALAT consensus recommends that the diagnosis of idiopathic interstitial pneumonias be based on the consensus between the clinician, radiologist and pathologist.2
Histopathological PatternIf the HRCT does not show a definitive pattern typical of UIP, the definitive diagnosis should be made by surgical lung biopsy.
Biopsies should be obtained in more than one lobe, and if possible, the middle lobe and lingula should be avoided, as they usually show non-specific changes that do not provide diagnostic information. Atelectasia due to the extraction can be minimized by gently instilling formaldehyde with a needle, or by shaking the tissue in the container with formaldehyde after removing the suture.
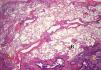
The histological pattern of UIP (Fig. 4) is defined by the fulfillment of four criteria: (a) evidence of marked fibrosis or distortion of the lung architecture, associated or not with honeycombing and predominantly subpleural and paraseptal; (b) presence of patchy lesions in which fibrotic areas are combined with areas of healthy lung; (c) presence of fibroblast foci in the areas of interface of fibrosis with healthy parenchyma and (d) absence of histopathological findings inconsistent with UIP. Among the characteristics not compatible with a UIP pattern are the presence of hyaline membranes, presence of foci with organizing pneumonia, granulomas, marked interstitial inflammatory cell infiltrate away from areas of honeycombing, predominantly airway centered changes, or the presence of other findings suggestive of an alternative diagnosis2 (Table 2).
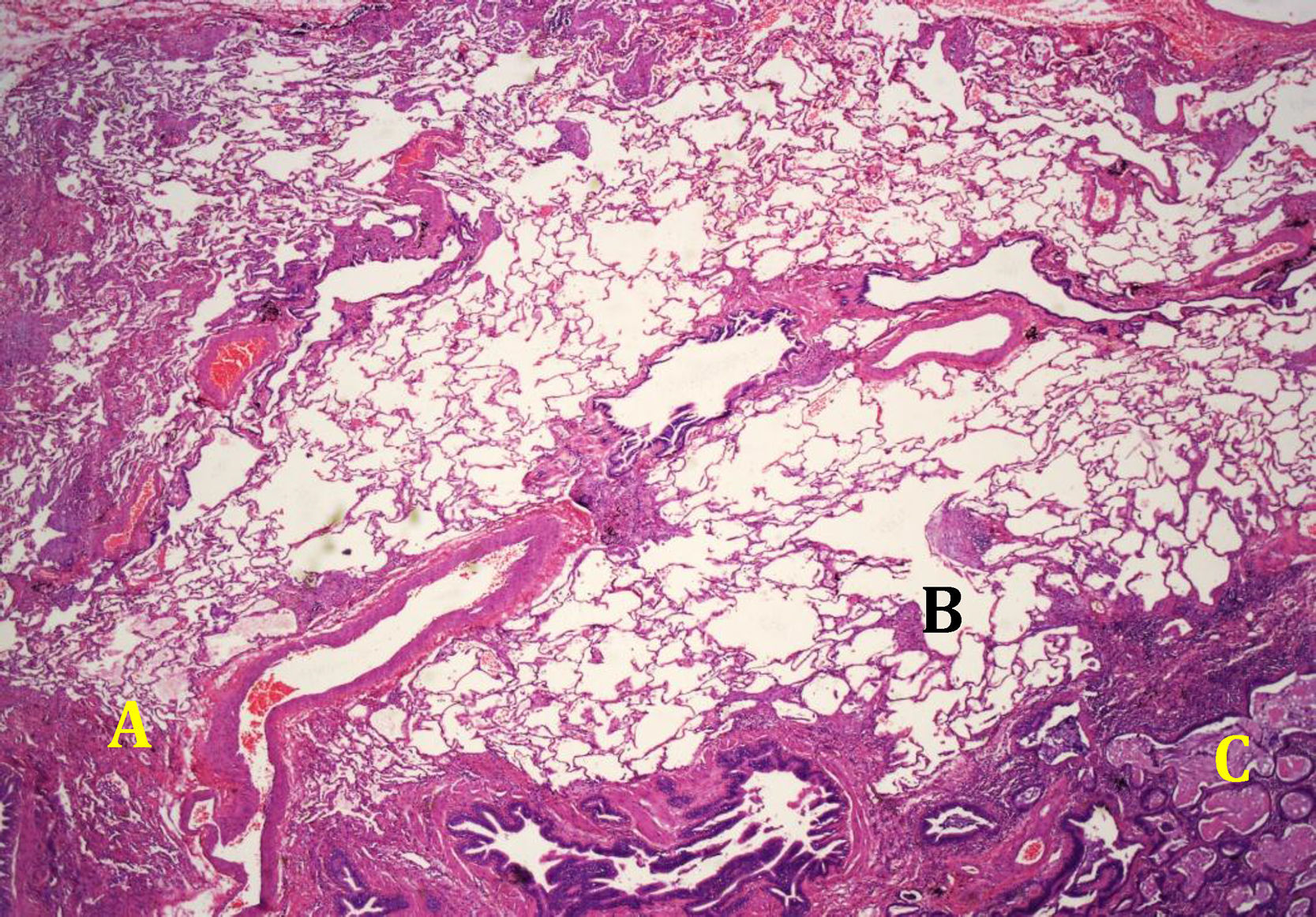
Usual Interstitial Pneumonia Pattern. Histopathological Criteria.
| UIP pattern (all four criteria) | Probable UIP pattern | Possible UIP pattern (all three criteria) | Not UIP pattern (any of the six criteria) |
| Evidence of marked fibrosis/architectural distortion, ±honeycombing in a predominantly subpleural/paraseptal distribution | Evidence of marked fibrosis/architectural distortion, ±honeycombing | Patchy or diffuse involvement of lung parenchyma by fibrosis, with or without interstitial inflammation | Hyaline membranesOrganizing pneumonia |
| Patchy involvement of lung parenchyma by fibrosis | Absence of either patchy involvement or fibroblastic foci, but not both | Absence of other criteria for UIP (see UIP Pattern column) | Granulomas |
| Presence of fibroblast foci | Absence of features against a diagnosis of UIP suggesting an alternate diagnosis | Absence of features against a diagnosis of UIP suggesting an alternate diagnosis | Marked interstitial inflammatory cell infiltrate away from honeycombing |
| Absence of features against a diagnosis of UIP suggesting an alternate diagnosis | Honeycomb changes only | Predominant airway centered changesOther features suggestive of an alternate diagnosis |
Adapted from Raghu et al.2
A histological pattern indistinguishable from UIP can be observed in systemic diseases (e.g. rheumatoid arthritis and scleroderma), chronic hypersensitivity pneumonitis, drug-induced pneumonitis, asbestosis and familial fibrosis, so the presence of granulomas, asbestos bodies, specific infections or other exogenous agents must be discarded in the biopsy. These are the reasons a UIP pattern should not be interpreted directly as an IPF pattern without first having excluded all these diseases. The integration of the HRCT findings with the histological pattern is used to establish the diagnosis of IPF, exclude it or, if the data are inconclusive, maintain it as probable or possible (Table 3).
Integration of HRCT Findings With the Histopathological Pattern.
| Histopathological pattern | |||||
| UIP | Probable UIP | Possible UIP | Non-classifiable fibrosis | Non-UIP | |
| HRCT pattern | |||||
| UIP | Yes | Yes | Yes | Yes | No |
| Possible UIP | Yes | Yes | Probable | Probable | No |
| Not consistent UIP | Possible | No | No | No | No |
UIP, usual interstitial pneumonia.
Adapted from Raghu et al.2
At the time of diagnosis, additional examinations recommended are assessment of the degree of cough and dyspnea, HRCT, respiratory function tests (forced spirometry, lung volumes, DLco), arterial blood gases, 6-minute-walk test and echocardiogram. In the follow-up of the progression, the examinations to be performed will depend on the patient's condition. In cases with mild pulmonary function abnormalities and absence of exercise limitation, the degree of cough and dyspnea should be evaluated, and respiratory function tests (FVC, DLco), resting pulse oximetry, chest radiograph (optional) and the 6-minute-walk test (6MWT) (optional) should be performed every 3–6 months. However, they should be conducted in shorter time periods if there are signs of disease progression. Measurement of total lung capacity (TLC) is not required to assess disease progression. If there is disease progression, blood gases, chest radiograph and the 6MWT should also be carried out sequentially, every 3 months, in order to evaluate the oxygenation and possibility of installing home oxygen therapy. It is not necessary to perform HRCT systematically and sequentially during follow-up. It should be carried out in cases of disease progression and suspected complications.2
There are various scales for assessing dyspnea intensity. The most widely used are the MRC (Medical Research Council), Borg scale, Baseline dyspnea index and Transition dyspnea index.24,25 The most useful method for assessing the cough intensity is the Leicester questionnaire.26 There are various questionnaires for assessing quality of life. The most widely used are the SF-36, St George Respiratory Questionnaire, and the University of California San Diego Shortness of Breath Questionnaire.27
Prognostic FactorsIPF is a disease with a variable clinical course, so it is important to identify factors that may help to define the patient prognosis. Various studies have evaluated clinical factors, biomarkers, radiological and physiological parameters (respiratory function tests and exercise capacity) and presence of comorbidities, associated with a higher risk of mortality, both at the time of follow-up and developmentally during follow-up.28 Factors associated with poor evolution are (Table 4):
- -
Aged over 70 years.28
- -
Associated comorbidities: pulmonary hypertension, emphysema and bronchogenic carcinoma.
- -
Degree of baseline dyspnea and its increase over time. The MRC dyspnea measurement scale has been shown to be very useful for determining disease progression.29
- -
DLco less than 40% (percentage of predicted value) at the time of diagnosis.30
- -
Decrease ≥10% in the FVC and ≥15% in the DLco (percentage of predicted value) in 6–12 months.30 The decrease in FVC is the lung function measure that best predicts mortality. Recently, du Bois et al.31 demonstrated that patients with a ≥10% decrease in FVC in 24 weeks have almost 5 times the risk of dying in the following year (HR 4.78; 95% CI 3.12–7.33), and those with a 5%–10% decrease in FVC have more than double the risk of death (HR 2.14; 95% CI 1.43–3.20). It is estimated that the minimal clinically important difference (MCID) in the variation in the FVC is 2%–6%. In the subgroup of patients with IPF and associated emphysema, in which the lung volumes are normal or slightly decreased, the variation in FVC does not predict survival.32
- -
Desaturation in the 6-minute-walk test. Both a peripheral arterial oxygen saturation (SpO2)≤88% and the distance covered are predictors of mortality in IPF.33 A reduction of >50m in the distance covered in 24 weeks is associated with a fourfold increase in the risk of death at one year (HR 4.27; 95% CI 2.57–7.10). The MCID is established at a distance of 24–45m.33 The desaturation discriminates which functional parameter is most suitable for the follow-up. In patients with SpO2≤88%, a decrease of >15% in the DLco in 6 months is the best predictor of mortality, while in those with SpO2>88%, the most significant parameter is a decrease of >10% in the FVC.34
The importance of heart rate recovery after the test has also been identified. A decrease of less than 14 beats after the first minute is an independent factor of poor prognosis. The 6MWT has good correlation with the maximum oxygen consumption (VO2max), measured in the cardiopulmonary exercise test. A VO2max value below 8.3mlkg−1min−1 is associated with a higher risk of mortality.35
- -
Extension of fibrosis on the HRCT (determined by the amount of reticulation, honeycombing and traction bronchiectasis) and its progression.36
- -
Biomarkers: although still not sufficiently validated to be included in patient follow-up, it has been reported that high serum levels of various proteins associated with the pathogenesis of IPF may predict mortality. These proteins include markers of alveolar epithelial cell injury (KL-6/MUC1, SP-A, MMP-7), alveolar macrophage activation (CCL-18), neutrophil recruitment and activation (S100A12, IL-8), and oxidative stress (ICAM-1, VCAM-1).37 High serum fibrocyte levels are associated with a poor prognosis and early mortality. High brain natriuretic peptide (BNP) concentrations are related with the presence of pulmonary hypertension and are a predictor of mortality in IPF.38 In patients in whom the diagnosis of IPF is made by surgical lung biopsy, the profusion of fibroblastic foci, quantified by semi-quantitative and quantitative methods, has also been shown to be a predictor of survival.
Predictive Variables of Survival.
| Baseline | Follow-up |
| Grade of dyspnea | Increase in grade of dyspnea |
| DLco≤40% | Decrease in FVC≥10% |
| Saturation<88% en 6MWT | Decrease in DLco≥15% |
| Extent of fibrosis on HRCT | Decrease >50m in 6MWT |
| Pulmonary hypertension | Worsening of fibrosis on HRCT |
| Biomarkers | Biomarkers |
DLco, capacity of lung carbon monoxide; FVC, forced vital capacity; 6MWT, 6-minute walk test; HRCT, high resolution computed tomography.
Multidimensional scales are currently being developed to try to predict the individual risk of mortality in patients with IPF. A study conducted using data from two clinical trials39 has established a clinical model composed of 4 factors (age, hospitalization, baseline FVC and change in 24 weeks) to predict the 1-year mortality risk. There are two recent studies, one retrospective40 using the GAP (Gender, Age, Physiology) index, which includes sex, age, FVC and DLco to predict the risk of mortality at 1, 2 and 3 years, and another prospective study29 which uses a risk stratification score (ROSE) to predict the 3-year survival, based on the degree of dyspnea (MRC scale), distance covered in the 6MWT and the CPI (composite physiological index, which includes FVC, FEV1 and DLco). There is insufficient evidence for the use of these scales in clinical practice.
Complications and ComorbiditiesPatients with IPF can develop complications and comorbidities that modify the clinical course and prognosis, either in relation to common physiopathogical mechanisms or as concurrent diseases associated with age.
Acute ExacerbationThis is defined as the rapid deterioration of the disease in the absence of infection, heart failure, pulmonary embolism or other identifiable cause. The diagnosis is based on the combination of clinical (worsening of baseline dyspnea in less than 4 weeks) and radiological findings (ground glass opacities or consolidations superimposed on a typical UIP pattern on the HRCT).41 The histological findings usually correspond to diffuse alveolar damage patterns, although they are sometimes characterized by organizing pneumonia or fibroblastic foci, added to the UIP morphological pattern. Its incidence and mortality vary according to the series, depending on the diagnostic criteria and follow-up time, with an incidence at 1 year of 9%–14% and at 3 years of 21%–24%,38 and a mortality of up to 60%–70% in 3–6 months. Various studies have established possible risk factors for the development of acute exacerbations, such as low FVC and DLco, and the presence of emphysema and pulmonary hypertension.42
Pulmonary HypertensionThe pulmonary hypertension that appears in IPF is included within the clinical group of PH associated with interstitial lung disease (group 3) of the updated Dana Point classification, and is defined as a mean pulmonary artery pressure (PaPm) greater than 25mmHg.43 Its prevalence is estimated at 30%–45% in patients evaluated for lung transplant, but it is much higher in those with longer disease evolution and at the time of transplant. The development of PH worsens the quality of life and functional status in these patients, who present more dyspnea, lower DLco and reduced exercise capacity, with a shorter distance covered and greater desaturation in the 6MWT.43 It is also associated with lower survival, with a mortality at one year of 28% in patients with PH compared to 5.5% in its absence.44 Although one study reported high pulmonary vascular resistance (PVR) with early mortality in IPF,45 it remains to be defined which hemodynamic parameter has most prognostic significance in these patients. Patients with IPF may present other comorbidities that also contribute to the development of PH, such as emphysema, sleep apnea–hypopnea syndrome, venous thromboembolism, heart disease and diastolic dysfunction.
Pulmonary EmphysemaCombined pulmonary fibrosis and emphysema (CPFE) is a syndrome with a diagnosis based on HRCT findings of centrilobular and paraseptal emphysema in the upper lobes and lesions compatible with UIP in lower lobes.46 Its prevalence is 30%–47% in patients with IPF and it usually appears in males with a history of smoking and major dyspnea. Spirometry and lung volumes are normal or minimally impaired (opposing effect between the hyperinflation due to the emphysema and the loss in volume due to the fibrosis), but there is a marked and disproportionate decrease in the DLco and significant exercise-induced hypoxemia (probably due to the additive effect of the emphysema, fibrosis and pulmonary vascular disease). In CPFE, the decrease in FVC and DLco is slower than in patients with IPF alone, and may cause an error in prognostic estimations. A recent study has demonstrated that the best predictor of mortality in these patients is the decrease in FEV1.47
In CPFE, precapillary PH appears more frequently, earlier and more pronounced than in patients with IPF without emphysema, which represents a poor prognosis and is the principal determinant of mortality. It remains to be determined whether CPFE represents a specific IPF phenotype in smokers, with different genetic predisposing factors and a different prognosis.
Gastroesophageal RefluxVarious studies have documented the high prevalence (66%–87%) of GER in patients with IPF, which is asymptomatic in most of them.48 The existence of GER and hiatus hernia, also more common in patients with IPF, may represent a factor in the pathogenesis and progression of the disease in relation to the presence of microaspirations.
Sleep Apnea–Hypopnea SyndromePatients with IPF have a high prevalence of sleep apnea-hypopnea syndrome (SAHS) and other breathing disorders during sleep.49 No correlation has been found between the severity of SAHS and physiological parameters such as FVC, DLco and lung volumes.49
Other ComplicationsThere is a higher risk of developing bronchogenic carcinoma in patients with IPF. The prevalence is 5%–10% and increases with the duration of the disease and in patients with CPFE.50 As well as smoking as a predisposing factor, various genetic mutations have been described that could be associated with its onset in these patients. An increased risk of cardiovascular diseases (heart disease and venous thromboembolism) is also found in patients with IPF.51 Pneumothorax can result in worsening of IPF. Its incidence is 11%.2
TreatmentBefore beginning treatment in patients diagnosed with IPF, the stage of the disease, prognostic factors and comorbidities must always be assessed. The therapeutic range includes52: (a) consider the currently available anti-fibrotic treatments, (b) avoid factors that aggravate the disease (gastro-esophageal reflux, respiratory infections, pulmonary hypertension, smoking), (c) treat the symptoms, mainly cough and dyspnea, (d) always have lung transplant in mind in cases that meet criteria, and (e) offer palliative treatment in the final phase of the disease.
Pharmacological TreatmentThe therapeutic approach in IPF began to change as of the new physiopathological hypothesis of the disease, in which the development of the condition was proposed as an epithelial–mesenchymal reparative abnormality that could commence without previous inflammation and where anti-inflammatory and immunomodulatory treatment had not been shown to modify the course of the disease.53,54 Based on this new concept, and following several advances in the understanding of the pathogenesis of pulmonary fibrosis, different research avenues were opened with the aim of inhibiting the fibrogenic process triggered, which was the beginning of the “anti-fibrotic” era. In the absence of other clinical therapeutic options, and despite the fact that the inefficacy of glucocorticoids in IPF had been known since 2003,53,55 the use of glucocorticoids±immunomodulators (azathioprine or cyclophosphamide)±N-Acetylcysteine was still considered in consensus guidelines as a treatment option. Table 5 specifies the recommendations and evidence for the pharmacological treatment of IPF.
Evidence-based Recommendations for the Pharmacological Treatment of Idiopathic Pulmonary Fibrosis.
| Agent | Mechanism of action | Recommendations |
| Recommended in selected patients | ||
| Pirfenidone | Antifibrotic+anti-inflammatory+antioxidant+anti-TGF β1 | Yes, weak recommendationa |
| NAC as monotherapy | Antioxidant | No, weak recommendation |
| Not recommended | ||
| Steroids+azathioprine+NAC | Immunosuppressant+antioxidant+anti-inflammatory | Do not use |
| Anticoagulation | Anticoagulant | Do not use |
| Bosentan | Dual endothelin-receptor antagonist | Do not use |
| Steroids as monotherapy | Immunosuppressant | Do not use |
| Steroids+immunomodulatory therapy | Immunosuppressant | Do not use |
| Colchicine | Inhibitor proliferation/collagen synthesis | Do not use |
| Cyclosporin A | Immunosuppressant | Do not use |
| Etanercept | Anti-TNF alfa | Do not use |
| Interferon gamma | Antifibrotic+immunomodulator | Do not use |
N-Acetylcysteine (NAC) increases the synthesis of glutathione, a potent antioxidant mediator, and decreases the fibrotic response in animal models of pulmonary fibrosis. A prospective, multicenter, phase III study (IFIGENIA study) evaluated the efficacy of NAC (1800mg/d) in a cohort of patients with IPF.56 The patients received NAC or placebo, combined with prednisone and azathioprine. After one year, patients who received NAC showed a lower rate of functional deterioration. The small number of patients included, absence of a placebo group and the short time period evaluated were widely discussed limitations. Nevertheless, the combination of glucocorticoid, azathioprine and NAC was the treatment of choice and recommended as a therapeutic option in consensus guidelines, until the results of the PANTHER study became known.57 The PANTHER study compared the efficacy of placebo vs NAC vs glucocorticoids combined with NAC and azathioprine. This study showed higher mortality and hospital admissions in patients who received the triple therapy compared to placebo or NAC treatment. Therefore, this triple therapy is not recommended. The trial is currently ongoing with only two arms: NAC and placebo. The true efficacy of NAC as monotherapy in the treatment of IPF will not be ascertained until the results become known.
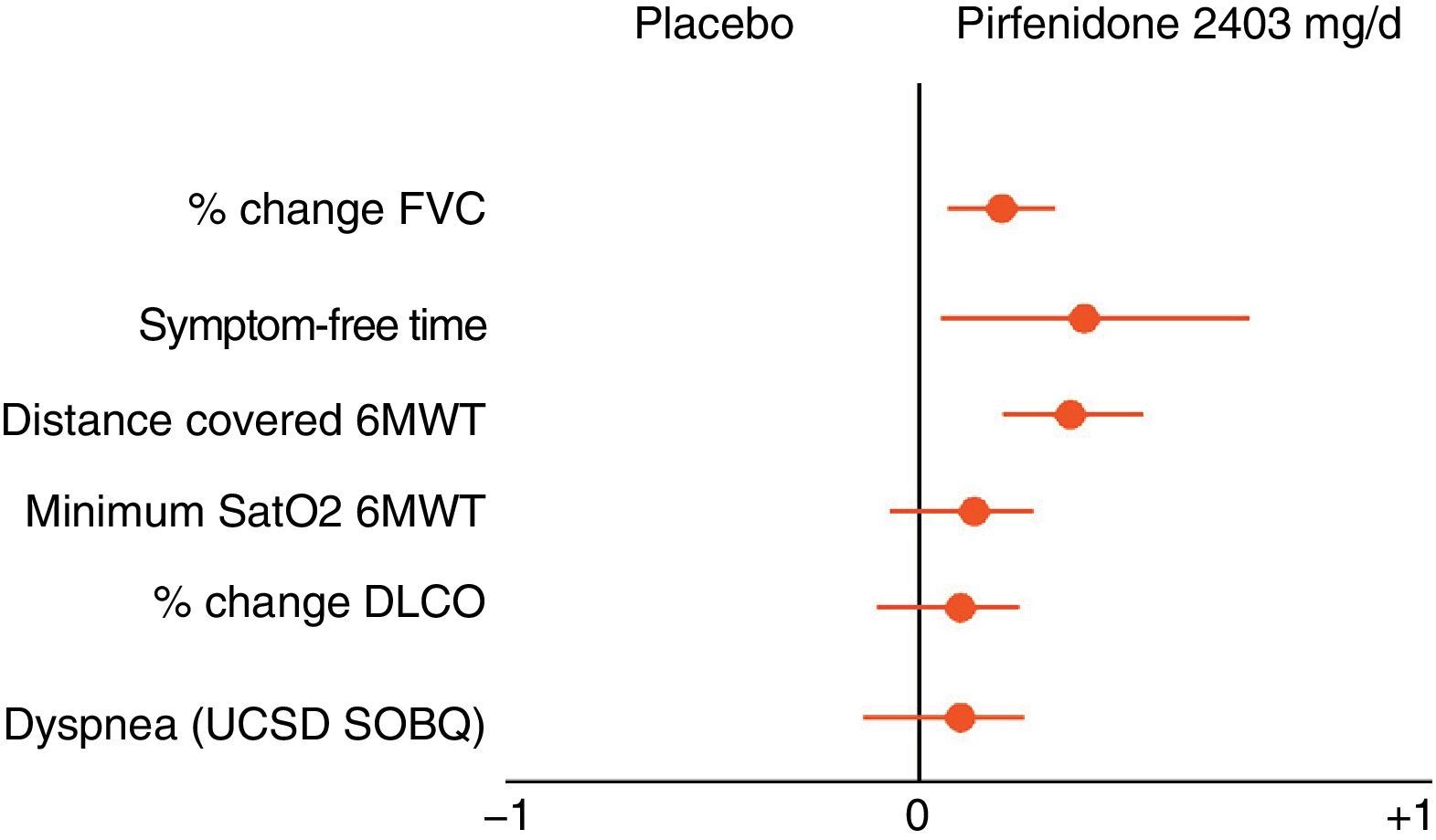
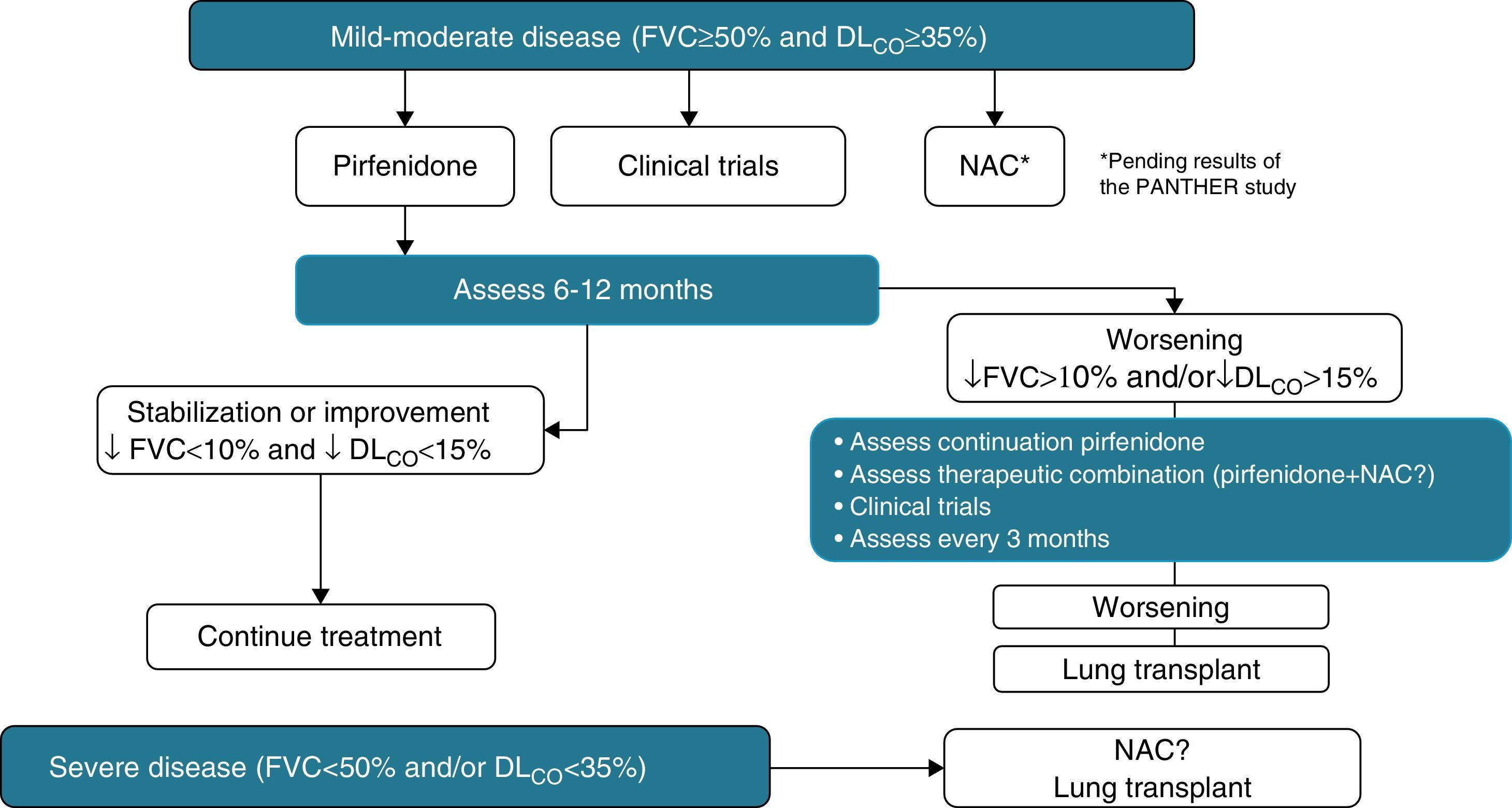
Pirfenidone (Esbriet®)Pirfenidone is a drug with anti-inflammatory and anti-fibrotic properties. It inhibits fibroblastic proliferation and the synthesis of pro-fibrogenic proteins and cytokines. Various experimental studies have demonstrated the anti-fibrotic effect of pirfenidone.58 Its clinical efficacy has been evaluated in three placebo-controlled, double-blind, randomized, multicentre, phase III studies in patients with IPF in Europe and the United States (CAPACITY studies) and Japan. The results of the CAPACITY studies have shown that pirfenidone, at doses of 2403mg/24h, reduces disease progression by 30%, and reduces the decline in FVC by 30%. Furthermore, a small percentage of patients presented rapid progression (20% pirfenidone with respect to 35% placebo) and a significant improvement was observed in the exercise capacity, determined by the distance covered in the 6MWT, as well as an increase in the progression-free interval (Fig. 5).55,59,60 To date, pirfenidone is the only drug with confirmed efficacy in the treatment of IPF. In March 2011, it was approved by the European Medicines Agency (EMA) for the treatment of mild-moderate IPF, defined as FVC>50% and DLco>35% (www.ema.europa.eu). At present, and as a result of the clinical trial results, its use is recommended in patients with FVC>50% and DLco>35%. Open-label studies are currently underway, in order to confirm the efficacy of the drug and to optimize its indications. The drug is available in several countries in the European Community, and will soon be available in Spain. It should be considered as a first line drug for the treatment of mild-moderate IPF (Table 4). The dose used is one 267mg capsule/8h for one week, 2 capsules/8h during the second week, and 3 capsules/8h from the third week onwards. The recommended treatment duration is at least 12 months. If there is improvement or stabilization of the disease, it seems logical that treatment should be continued. In the case of deterioration, the advisability of continuing treatment or instigating other therapeutic strategies should be considered for each patient. In this context, it should be pointed out that there are no data showing that the addition of NAC increases the efficacy of pirfenidone (Fig. 6). The main contraindications are drug hypersensitivity, concomitant use of fluvoxamine, severe liver disease or nephropathy, and pregnancy (its use in this population has not been confirmed). Pirfenidone interacts with omeprazole, so it is advisable to substitute this drug for pantoprazole. The most common side effects are dizziness, photosensitivity (skin and eye protection is essential), stomach upsets (administer the drug with food) and liver function abnormalities, generally reversible on reducing the dose.61
Nintedanib is a potent tyrosine kinase inhibitor that acts on the vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF) and fibroblast growth (FGF) receptors. The results of the TOMORROW phase II clinical trial have shown that the administration of BIBF-1120 150mg/12h resulted in a positive trend toward a reduction in the loss of FVC and in exacerbations of the disease, and improvement in quality of life.62 A phase III clinical trial is currently underway to assess its efficacy in the treatment of IPF.
Other Anti-fibrotic DrugsDuring the last decade, various randomized phase II and III clinical trials have been conducted with drugs experimentally considered as anti-fibrotics, but which have not demonstrated any efficacy for the treatment of IPF.63 These include imatinib mesylate (Gleevec), interferon gamma 1-beta, tumor necrosis factor-α antagonists (etanercept), anticoagulant (warfarin), endothelin antagonists (bosentan, macitentan, ambrisentan) and sildenafil (Table 4). Furthermore, phase I and II clinical trials with other molecules are ongoing (www.clinicaltrial.gov): anti-IL13/–IL4 antibody (SAR156597), angiotensin-II AT1 receptor antagonist (losartan), anti-integrin αvβ6 monoclonal antibody (STX-100), collagen V (IW-001), anti-IL13 antibody (QAX576), connective tissue growth factor antagonist (FG-3019), anti-LOXL2 or GS-6624 antibody (AB0024), pentraxin-2 or rhPTX-2 recombinant protein (PRM-151) and sirolimus. Finally, there are infinite experimental options considered as possible anti-fibrotics. These include anti-TGF-β1 monoclonal antibody CAT-192, hepatocyte growth factor (HGF), prostaglandin-E2 (PGE-2) and interfering RNAs (nkRNA and PnkRNA). In the future, effective anti-fibrotic therapy for IPF could include a combination of drugs that act synergically on different pathogenic pathways of the disease.
Treatment of Complications and ComorbiditiesThe complications of IPF and the co-existence of comorbidities play a determining role in the course of the disease, hence the importance of their early detection and treatment. The most relevant conditions due to their severity and prognostic implications are acute exacerbation, pulmonary hypertension and gastroesophageal reflux.2
Acute ExacerbationThere are no randomized, controlled trials to date that support a certain treatment for the acute exacerbation of IPF, so current recommendations are based on the results of the few series published, most retrospective or including few patients.64 The most widely used treatment is high-dose corticoid boluses (methylprednisolone 500–1000mg/day) for 3 days, followed by high-dose prednisone (0.5mg/kg/day), which are gradually reduced, combined or not with immunosuppressants such as azathioprine, cyclophosphamide or cyclosporine (weak recommendation, very low quality evidence). Together with this treatment, the consensus among experts defends support treatment, similar to that in acute respiratory distress syndrome65 (weak recommendation, very low quality evidence). Several studies have shown that mechanical ventilation in patients with IPF and respiratory failure is not effective in most cases.66
Pulmonary HypertensionThe use of sildenafil has been evaluated in two non-placebo controlled randomized trials which showed an improvement in the exercise capacity in patients with pulmonary hypertension associated with IPF (weak recommendation, very low quality evidence).67,68 The few studies conducted with epoprostenol or bosentan in patients with pulmonary hypertension associated with DILD (only some had IPF) did not allow definitive conclusions to be drawn with respect to the generalized indication for their use in patients with pulmonary hypertension associated with IPF. Therefore, the current recommendation is that patients with moderate-severe pulmonary hypertension demonstrated by right catheterization (PAPm>35mmHg) may be candidates for treatment with vasomodulator drugs such as sildenafil (weak recommendation, very low quality evidence).
Gastroesophageal RefluxGastroesophageal acid reflux is a risk factor for aspiration, a recognized cause of pneumonitis, so it could contribute to chronic inflammation of the airways and also to fibrosis. In this, clinical stabilization of abnormal gastroesophageal reflux, once treated either by drugs or surgical procedures, has been documented.48 Given the reasonable cost and low morbidity that could result from the side effects induced by proton pump inhibitors, it is reasonable to prescribe them in patients with IPF and proven gastroesophageal acid reflux, together with anti-reflux measures (weak recommendation, very low quality evidence).
Non-pharmacological TreatmentHome Oxygen TherapyThe indication for home oxygen therapy in patients with IPF and resting hypoxemia comes, basically, from extrapolating the conclusions of studies conducted in patients with COPD and chronic respiratory failure. There are no conclusive data supporting the use of ambulatory oxygen therapy for patients who desaturate only during exercise, with no respiratory failure at rest.69 Two recently published retrospective studies indicate that home oxygen therapy could improve performance in the 6MWT in patients with DILD.70,71 Recently, a cohort study on 104 patients with IPF and 151 patients with other DILD found that the major requirement for oxygen therapy, i.e. the highest FiO2 necessary to maintain a basal SaO2≥96% before performing the 6MWT, is an independent risk factor associated with mortality in patients with IPF after one year of follow-up (consistent recommendation, very low quality evidence).72
Due to the lack of specific data in patients with IPF, it is recommended that long-term home oxygen therapy be administered when there is a finding of significant hypoxemia at rest or in the 6MWT (SaO2≤88%) (consistent recommendation, very low quality evidence).
Lung TransplantLung transplant is the only treatment for advanced IPF that results in a major functional improvement and an increase in the 1, 5 and 10-year survival of 74%, 45%, and 22%, respectively.73 These rates are significantly lower than those observed in patients who have undergone transplants for other respiratory diseases, such as alpha-1-antitrypsin deficiency, pulmonary hypertension, cystic fibrosis or COPD, but although the results of transplantation in patients with IPF are worse, for the time being it is the only effective therapeutic alternative, still exceeding the results of the best pharmacological treatment available. Therefore, IPF patients with progressive disease should be evaluated in a lung transplant unit, regardless of the type of medical treatment that they receive, providing that there are no contraindications for the surgical procedure (consistent recommendation, low quality evidence).73
Respiratory RehabilitationThe 2008 Cochrane Collaboration review updated in 2010 confirmed that rehabilitation is safe in patients with DILD (and also in the subgroup of patients with IPF) in terms of improving the distance covered in the 6MWT and health-related quality of life.74 However, the beneficial effects of rehabilitation on long-term survival are not documented. Recent studies with follow-up at 6 months show that patients with IPF obtain more lasting benefits from rehabilitation programs when the disease is mild, while the other DILD obtain them regardless of the severity of the disease.75,76 It would be advisable to include patients with IPF in a respiratory rehabilitation program before the disease reaches advanced stages (weak recommendation, low quality evidence).
Cell Therapy and Gene TherapyAt present, both cell therapy and gene therapy in IPF are in experimental study phases, so we will still have to wait some time to learn their effectiveness as an alternative treatment or combined with pharmacological treatments.
IPF is characterized by the death of alveolar epithelial cells, which are replaced by fibroblasts. Therefore, therapeutic approaches based on cell therapies are aimed at replacing the alveolar cells responsible for regenerating the alveolar epithelium. During the last decade, the transplant of stem cells with the ability to proliferate and differentiate into alveolar cells has been proposed as a therapeutic strategy. Mesenchymal stem cells are probably the cells that have been most widely studied and used, taking advantage of their capacity to differentiate into numerous different cell types. The results obtained on administering mesenchymal cells are contradictory. On the one side, it has been observed that they are capable of adhering to the alveolus and adopting an alveolar cell phenotype, but on the other, it has been reported that, contrary to expectations, they differentiate into fibroblasts, and thus would be increasing the intensity of the fibrosis rather then reducing it. This raises doubts about whether the administration of mesenchymal cells could be considered an ideal therapy in patients with IPF.77
To date, the best option proposed as cell therapy has been the administration of alveolar type II cells, which have managed to reverse the fibrogenesis process in an animal model of bleomycin-induced pulmonary fibrosis.78 These results indicate that the administration of alveolar type II cells could become a treatment for IPF. However, this will involve waiting for the results of currently ongoing clinical studies.
Although IPF is not a genetic disease, there are numerous studies showing that certain genetic polymorphisms may be associated with a higher susceptibility to developing IPF.8 Like cell therapies, gene therapy has only been carried out at experimental level, basically by inhibiting or administering different microRNAs that regulate the expression of different genes related with IPF.
Palliative CareThe treatment of cough (especially nocturnal cough which makes it difficult to sleep) and dyspnea is a determining factor for maintaining an acceptable quality of life in patients with IPF. Codeine and other opiates and low-dose glucocorticoids (prednisone 5–10mg/day) have shown certain efficacy in the control of cough.78
In a clinical trial, it has been shown that thalidomide improves cough and respiratory quality of life (consistent recommendation, moderate quality evidence).79 Low doses of morphine can improve both the sensation of dyspnea and persistent cough in patients with advanced disease.78
Palliative care should be aimed at improving the quality of life of patients and their families facing the problems inherent in this progressive and so far incurable disease. Thus, the early identification, assessment and treatment of symptoms such as pain, dyspnea and uncontrollable coughing, as well as any other symptom related with disease progression, in both the physical and psychosocial sphere, is important.80 It is therefore recommended that palliative care be considered an integral part of the overall treatment of IPF (consistent recommendation, very low quality evidence).
Conflict of InterestsAntoni Xaubet declares that he has received funding for giving lectures at educational events and/or for scientific advice and/or research from Intermune, Actelion, Almirall and GSK in relation to the subject of the guidelines.
Julio Ancochea declares that he has received funding for giving lectures at educational events and/or for scientific advice and/or research from Boehringer Ingelheim, Intermune and Zambon in relation to the subject of the guidelines.
Anna Serrano-Mollar declares that she has received funding for giving lectures at educational events and/or for scientific advice and/or research from Intermune in relation to the subject of the guidelines.




www.publicationethics.org.




Archivos de Bronconeumología follows the Recommendations for the Conduct, Reporting, Editing and Publication of Scholarly Work in Medical Journals