



Introduction
Endomyocardial fibrosis (EMF) is a rare form of restrictive cardiomyopathy that is more common in tropical countries. It is characterized by a proliferation of fibrous endocardial tissue in one or both ventricles especially around the apex and inflow tract, with consequent diastolic malfunction, and it can be the cause of thromboembolic events.1 Although it is not unusual to find thrombi covering endocardial lesions, it is exceptional to come across chronic thromboembolic pulmonary hypertension (PHT) caused by recurrent emboli. Very few cases are described in the literature.2
We report a case of chronic thromboembolic PHT in a 51-year-old woman who was diagnosed with EMF and who had undergone surgery 20 years earlier.
Clinical observations
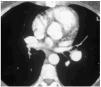
The patient was a 51-year-old woman from Dutch Guyana. She was a non-smoker, had no other toxic habits, had been operated on 20 years earlier for signs consistent with EMF and had attended regular check-ups with a cardiologist. She was referred from another hospital for evaluation of PHT detected by an echocardiogram during a routine visit. The patient had experienced worsening dyspnea over the previous few years. Auscultation revealed only an increase in the second heart sound, other sounds being normal. A chest x-ray showed several prominent pulmonary hila that seemed to correspond to blood vessels. An electrocardiogram confirmed a right bundle branch block. A complete blood work-up gave normal counts, including for antinuclear antibodies, extractable nuclear antigen antibodies, and anti-neutrophilic cytoplasmic antibodies. A Doppler ultrasound scan of the lower extremities showed no signs of deep venous thrombosis. Echocardiography showed dilation and hypertrophy of the right ventricle with calcification of the subvalvar apparatus of the tricuspid valve and the papillary muscles, as well as moderate to severe PHT with systolic pressure in the pulmonary artery estimated at 75-78 mm Hg. An echocardiograph carried out in 1981 showed pulmonary artery pressure (PAP) to be 30 mm Hg.
A lung ventilation-perfusion scan was requested (Figure 1) and showed multiple perfusion defects in both lungs in well-ventilated zones. The presence of pulmonary thromboemboli was therefore considered highly probable. Contrast-enhanced helical computed tomography (CT) confirmed dilation of the pulmonary artery and its branches as well as repletion defects in the right interlobar artery (Figure 2), which was reduced in diameter. Right heart catheterization revealed a systolic PAP of 65 mm Hg, a diastolic PAP of 30 mm Hg and a mean PAP of 45 mm Hg. After administering inhaled iloprost, a prostacyclin (prostaglandin I2) analogue, mean PAP fell 20%, with a resulting reduction in systemic blood pressure of the same amount. As none of the hospitals to which the patient could be referred carried out pulmonary thromboendarterectomy the patient continued to receive treatment with oral anticoagulants and inhaled iloprost.
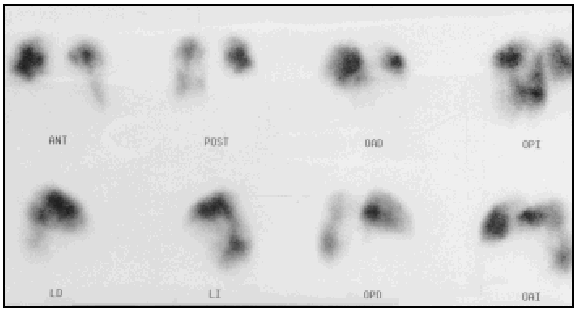
Fig. 1. A lung ventilation-perfusion scan showing multiple segmentary perfusion defects in both lungs.
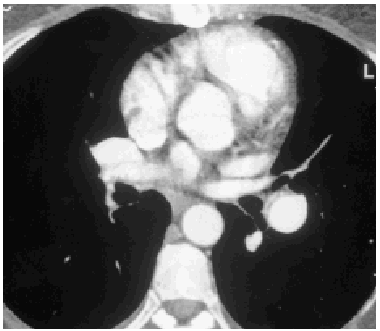
Fig. 2. Helicoidal CT of the thorax showing reduced diameter of the right interlobar artery.
Discussion
EMF is an endemic illness in tropical and subtropical countries. First described in Brazil in 1954 but rare in the Spanish climate,3,4 it is a type of restrictive cardiomyopathy caused by fibrosis of the endocardium that sometimes extends to the subjacent myocardium. Fibrosis may occur in the right or left ventricle, or both, is usually situated at the inflow tract or the apex, and sometimes affects the papillary muscles and the chordae tendineae. Because the prevalence of EMF is high in tropical countries that have frequently occurring parasitic illnesses and associated eosinophilia, a toxic effect of eosinophils on the heart has been postulated as the pathogenic mechanism. Other recent hypotheses link EMF with the high dietary cesium levels and low magnesium levels common in such countries.5 Clinical course depends on the degree of ventricular fibrosis and the seriousness of concomitant mitral/tricuspid valve insufficiency. The best treatment for patients with these symptoms is surgery involving endocardial decortication and valve repair or replacement, which has a 17-year survival rate of 55% in some series.6
Diagnosis is based on clinical observations and above all on echocardiographic findings. Endocardial calcification has been described but is hardly ever found.7 It is not unusual to find mural thrombi, sometimes calcified, covering the endocardial lesions and giving rise to emboli.8 When EMF only affects the right ventricle (in 10% of patients) the silent and repeated dislodgement of these thrombi can give rise to chronic thromboembolic PHT.2
Chronic thromboembolic PHT is a rare sequela of untreated or recurrent pulmonary embolism with the organization and incorporation of the thrombi into the pulmonary artery walls, arterial occlusion and subsequent recanalization.9 Clusters of thrombi fuse with the endothelium and become part of the lining of pulmonary blood vessels, causing a rise in vascular resistance and PAP. Recent studies show that pulmonary emboli can occur without producing symptoms or may be diagnosed incorrectly, and for this reason patients may remain asymptomatic for months or years.10 The most sensitive noninvasive diagnostic method is still the pulmonary ventilation-perfusion scan, which invariably records one or more defects affecting segments or even lobes in chronic thromboembolic PHT patients, whereas normal results or only sub-segmentary defects are seen in cases of primary PHT.9 In a study analyzing 25 patients with PHT the authors showed that 8 patients with chronic thromboembolic PHT had high probability scans while 17 patients with primary PHT had normal or low probability scans.11 Other causes of segmentary defects on the lung scan, such as pulmonary artery sarcoma, extrinsic vascular compression by benign tumors or fibrosis of pleural tissues, or vasculitis of the large pulmonary blood vessels are rarer and were ruled out in our patient. The role of helical and enhanced contrast CT in diagnosing patients with chronic thromboembolic PHT remains to be established; while such imaging may reveal a variety of abnormalities such as collateral flow in the bronchial arteries, thrombi in the central pulmonary arteries, or changes in the mosaic pattern of the pulmonary parenchyma, none of these findings is specific to the illness.12 The definitive method of diagnosis is still pulmonary angiography, which is especially useful for defining the exact position and size of the thrombus when considering the possibility of surgery (thromboendarterectomy), although that diagnostic procedure is not exempt from hemodynamic complications in cases of severe pulmonary hypertension.
Pulmonary thromboendarterectomy, the definitive treatment for chronic thromboembolic PHT, is a complex procedure that is used very little in Spain (performed only in one hospital as far as we know) and requires partial sternotomy, cardiopulmonary bypass, deep hypothermia and periods of hypothermic circulatory arrest to ensure adequate visibility for the dissection and removal of thrombotic material.13 Approximately 30% of patients about to undergo thromboendarterectomy first undergo pulmonary angiography. This is done using a fiberoptic catheter 120 cm long and 3 mm in external diameter inserted through the right internal jugular vein into the pulmonary artery under fluoroscopic guidance. An angioplasty balloon is then inflated with carbon dioxide to stop the blood flow and allow a view of the artery walls. The six-year survival rate in a series of 532 patients who underwent such surgery was 75%.14
At present several treatments are able to improve life expectancy in patients with various kinds of PHT: oral anticoagulants, high dosages of calcium antagonists, and inhaled prostacyclin.15 When patients with chronic thromboembolic PHT are considered ineligible for thromboendarterectomy, because of difficult surgical access or poor functional reserve, or because facilities are unavailable for surgery as in our case, possible treatments for which preliminary reports of improvement have been published include pulmonary vasodilation with iloprost or a combination of iloprost and sildenafil;16,17 only short-term results of such treatments are known at this time, however. Other more sophisticated techniques such as balloon pulmonary angioplasty have been shown to give good results, but in a very small number of patients.18
Correspondence: Dr. E. Fernández Vázquez.
Servicio de Neumología. Hospital Universitario Virgen de las Nieves.
Avda. Fuerzas Armadas, s/n. 18014 Granada. Spain.
E-mail: efernandezv@hvn.sas.junta-andalucia.es
Manuscript received December 12, 2002.
Accepted for publication February 3, 2003.







