

IgG4-related disease is a multisystem disorder characterized by the formation of fibroinflammatory lesions, causing the affected tissues to malfunction.1
The 2 main features of this disease are tissue infiltration by IgG4 and raised IgG4 levels in serum, but these phenomena are not present in all patients.2
IgG4-related disease represents a diagnostic challenge as it mimics many other processes, including cancers, infections, and autoimmune diseases. For this reason, in most cases, the affected tissues must be biopsied in order to arrive at a diagnosis.
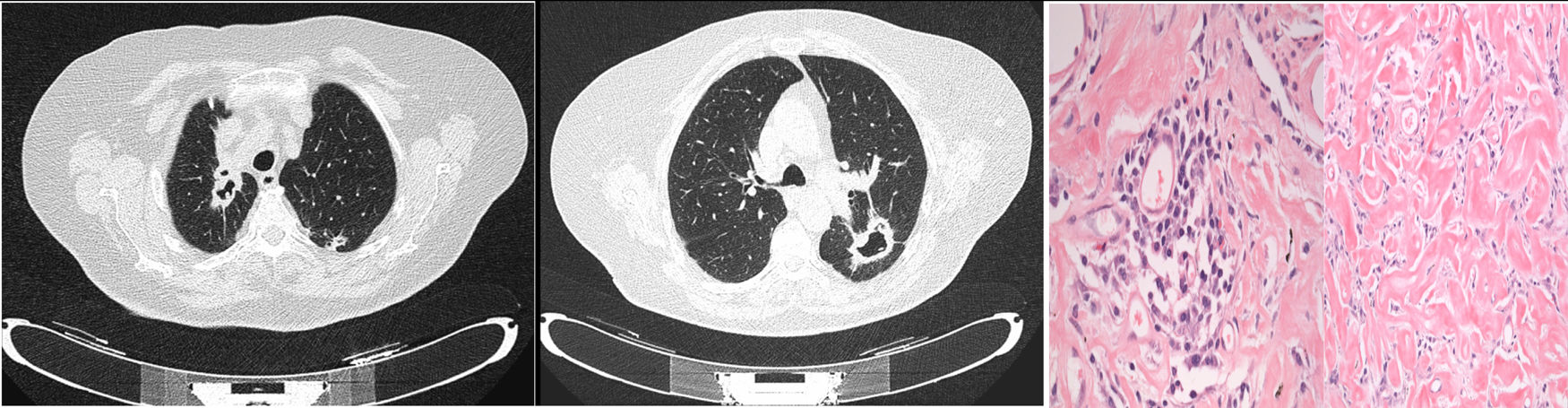
We report the case of an asymptomatic 79-year-old woman, former smoker, who was referred to our department for the study of lung nodules, detected incidentally on a chest X-ray obtained for a preoperative work-up. Fiberoptic bronchoscopy showed no significant changes. Chest computed tomography (CT) (Fig. 1) was performed, which revealed 2 lesions of thick irregular morphology with large central cavitation and coarse peripheral calcifications lacking interior content in both apical segments, measuring 45 and 40mm in diameter in the right upper lobe (RUL) and left upper lobe (LUL), respectively.
Clinical laboratory testing showed only positive antinuclear antibodies (ANA), titer 1/160 with a granular pattern, and normal IgG4 levels (53mg/dl).
Left thoracotomy was performed with a wedge resection of LUL segment 6 and vertex which included a nodular lesion measuring 6×3×1.7mm. An inflammatory infiltrate with abundant plasma cells (Fig. 1) was observed that was diagnostic for nodular pulmonary disease due to IgG4-deposition disease (the immunohistochemical study expressed abundant IgG with a IgG4/IgG ratio >40%).
Taking into account that the patient remained asymptomatic at all times, and that her IgG4 subclass levels were normal, we decided to continue with clinical and radiological follow-up. After 3 years of follow-up, the patient remains asymptomatic and her radiological lesions are stable.
IgG4-related disease is a recently described condition that more often affects men (ratio of 1 to 0.7) between 60 to 65 years old, predominantly individuals of Asian race.1 It is characterized by raised serum IgG4 and IgG4 infiltration in the affected tissue.2
In 1995, Yoshida et al.3 described a form of chronic pancreatitis and postulated that an autoimmune mechanism was the cause of the pancreatic lesion. However, it was not until 2003 that Kamisawa et al.4 first established the term “IgG4-related autoimmune disease” (formerly also known as “hyper-IgG4 disease” and “sclerosing disease”), while demonstrating that patients who had autoimmune pancreatitis due to this mechanism could also have extensive lesions in other tissues.
Associated symptoms depend on the organ involved and include abdominal pain (40%), respiratory symptoms (13%), pruritus (13%), and diarrhea (6%). Constitutional symptoms such as weight loss, fatigue and low-grade fever may occur. In addition, some subjects can be asymptomatic at the time of diagnosis and show abnormalities only in laboratory tests or imaging studies.
Patients with intrathoracic involvement are often asymptomatic, and are typically diagnosed after an incidental finding on an imaging study. Approximately 38% of patients have respiratory symptoms, usually cough and dyspnea on exertion. The most frequent intrathoracic manifestation is the presence of hilar and mediastinal lymphadenopathies (40%–100%). The 2 most common forms of pulmonary parenchymal involvement are rounded opacities (nodules or masses) and interstitial lung disease.5
Serologically, peripheral eosinophilia is found in 34% of patients. Most patients with intrathoracic involvement have raised IgG4 levels in peripheral blood (>140mg/dl), but this biomarker is considered to be relatively insensitive and nonspecific for the diagnosis of this disease.6 The usefulness of other immunological markers such as rheumatoid factor or C-reactive protein is limited.
Information from bronchoalveolar lavage (BAL) in IgG4-related lung disease (predominance of lymphocytes with a normal CD4+/CD8+ ratio) is scant and its role is yet to be defined.
Histopathological findings are needed for the diagnosis of the disease. The specimen of choice is surgical lung biopsy, but transbronchial biopsy can also be helpful. Eosinophilic infiltration in observed the interlobular and peribronchial septa and the presence of granulomas is rare.7 In the lung, collagenized fibrosis and active fibroblast proliferation are more predominant than in autoimmune pancreatitis.
The diagnosis of IgG4-related disease is, then, based on a combination of histopathological, clinical, serological and radiological characteristics.8
Steroid treatment is the modality most often recommended in the literature, and the response of intrathoracic and extrapulmonary lesions is similar. The optimal dose is not known, but most experts recommend oral prednisone at an initial dose of 30 to 40mg/day,9 while 1mg/kg/day can be used if progress is slow. These doses must be maintained for 2 to 4 weeks and may be reduced in the case of a good response, but patients must always be closely monitored for possible relapse. The optimal duration of maintenance therapy has not been determined and some patients with pulmonary involvement may not achieve complete resolution of their disease.
Evidence on the use of immunosuppressive therapy other than corticosteroids is scant.9 Treatment with rituximab in combination with steroids has shown promising results in retrospective studies, and in an open-label pilot study10 this combination even demonstrated a favorable disease response.
The prognosis of this disease depends on several parameters, including factors specific to both the patient and the disease. It must also be borne in mind that disease relapse after the withdrawal of steroids is relatively common.
Please cite this article as: Aguado Ibáñez S, et al. Nódulos pulmonares cavitados con relación a la enfermedad por depósito de IgG4. Arch Bronconeumol. 2020;56:127–128.







