


Kartagener's syndrome is a rare autosomal-recessive genetic disease with progressive damage of the respiratory system and situs inversus. Although the management of patients with Kartagener's syndrome remains uncertain and evidence is limited, it is important to follow up these patients with an adequate and shared care system. This report presents a clinical case of Kartagener's syndrome in a 25-year-old woman. Computed tomography showed dextrocardia and bronchiectasis. Abdominal X-ray and ultrasound confirmed situs inversus totalis. After 7 years, good treatment results were achieved: lung function improved and radiological findings showed no changes. The present case discusses the complex interrelationship between the genetic variation and a proper nonspecific management of Kartagener's syndrome.
El síndrome de Kartagener es una enfermedad genética poco frecuente que se hereda de forma autosómica recesiva, con una afectación progresiva del sistema respiratorio y situs inversus. Aunque el tratamiento de los pacientes con el síndrome sigue siendo poco claro y las pruebas disponibles son limitadas, es importante su seguimiento con una asistencia adecuada y compartida. En el presente informe se describe un caso clínico del síndrome en una mujer de 25 años de edad. La tomografía computarizada demostró dextrocardia y bronquiectasias. La radiografía simple y la ecografía abdominal confirmaron un situs inversus total. Después de 7 años, se obtuvieron resultados satisfactorios del tratamiento: la función pulmonar mejoró y en la exploración radiológica no se demostraron cambios. En el presente artículo se describe la compleja interrelación entre la variación genética y un tratamiento inespecífico apropiado del síndrome.
Kartagener's syndrome is a recessive autosomal disease which is mainly seen to affect ciliary movement.1 The syndrome forms part of a larger group of diseases that are referred to as primary ciliary dyskinesia (PCD). Although the disease is inherited in a recessive autosomal pattern and some specific genetic defects have been recognized, the syndrome clearly manifests substantial genetic heterogeneity.2 The incidence of the process is 1–2/30000 births.3 Manes Kartagener, a pulmonologist in Zurich, described the triad of sinusitis, bronchiectasis and situs inversus for the first time in 1933.4 It is interesting to note that Dr. Kartagener's surname is Sephardi (Spanish Jew) and derived from the name of the Spanish city of Cartagena, which at the same time derives from the name of the Phoenician city of Carthage.4
The symptoms of the syndrome are a consequence of the defective motility of the cilia found in the respiratory tract.2,3 Recurrent lung infections are due to the affected mucociliary movement in the airways, which cause mucus stasis in the bronchi.1,5 Until a diagnosis is reached, progressive and substantial lung damage may occur.3,5 In older children and adults with primary ciliary dyskinesia, 3 diseases of the lower respiratory tract have been described: pneumonia, bronchiectasis and asthma.6 Although treatment for patients with this syndrome has yet to be established, it is important to control chronic lung infections and declining lung function. The efforts made at identifying interactions between genetic factors and environmental determinants could translate into greater knowledge about the pathogenesis of the bronchiectasis.
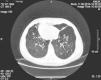
Case ReportWe present a clinical case of Kartagener's syndrome in a 25-year-old woman. The patient complained of cough and expectoration. An examination of the respiratory system revealed coarse crackles with rhonchus throughout the thorax. The apex beat was located on the right side, and the liver was palpable in the left hypochondria. Computed tomography (CT) of the chest demonstrated dextrocardia and bilateral bronchiectasis (Fig. 1). Sinus radiographs showed inflammation of the nasal mucous and mucosal hypertrophy of the maxillary sinuses, indicative of chronic rhinosinusitis. Mucociliary clearance was assessed with the saccharin test, with a time of 48min (normal, <15min). Spirometry demonstrated mild obstruction: expiratory volume in the first second (FEV1), 93% of the reference value (3.27 l); forced vital capacity (FVC), 110% (4.40 l); FEV1/VC, 74%. We analyzed other genetic markers that are important for pulmonary protection: alpha-1-antitrypsin (AAT), antiprotease and secretory immunoglobulin A (IgA). The AAT genotype of the patient was the wild type (PiMM) and the concentration of AAT was found in the upper limit of the normal range, in 2g/l. The level of immunoprotective IgA was also high, at 3.5g/l.
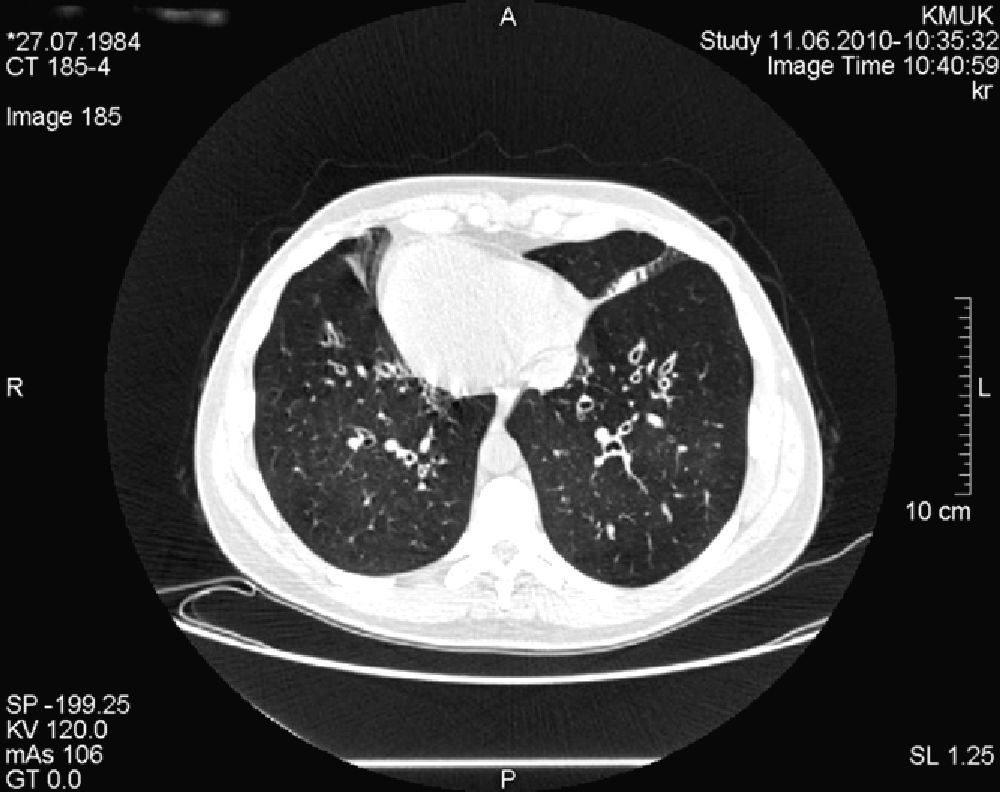
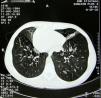
The pathological history of the patient included repetitive lung infections, chronic sinusitis and chronic otitis since early childhood. At the age of 14, she was diagnosed with Kartagener's disease. The diagnosis was based on clinical symptoms and radiology. The screening exam of the patient's family genealogy did not identify other cases with the syndrome. At the time of diagnosis, spirometry demonstrated bronchial obstruction of the first order bronchi: FEV1, 88% of the reference value (3.08 l); FVC, 111% (4.45 l); FEV1/CV, 69%. Starting at the age of 18, the patient had been followed by pulmonologists and geneticists. Chest CT (Fig. 2) revealed dextrocardia, lung fibrosis and multiple bronchiectasis in the upper lobes. Plain radiography and abdominal ultrasound confirmed situs inversus totalis (left liver, gastric bubble and spleen on the right). On the microbiologic examination, the most common isolated pathogens were Haemophilus influenzae, Pseudomonas aeruginosa and Serratia marcescens.
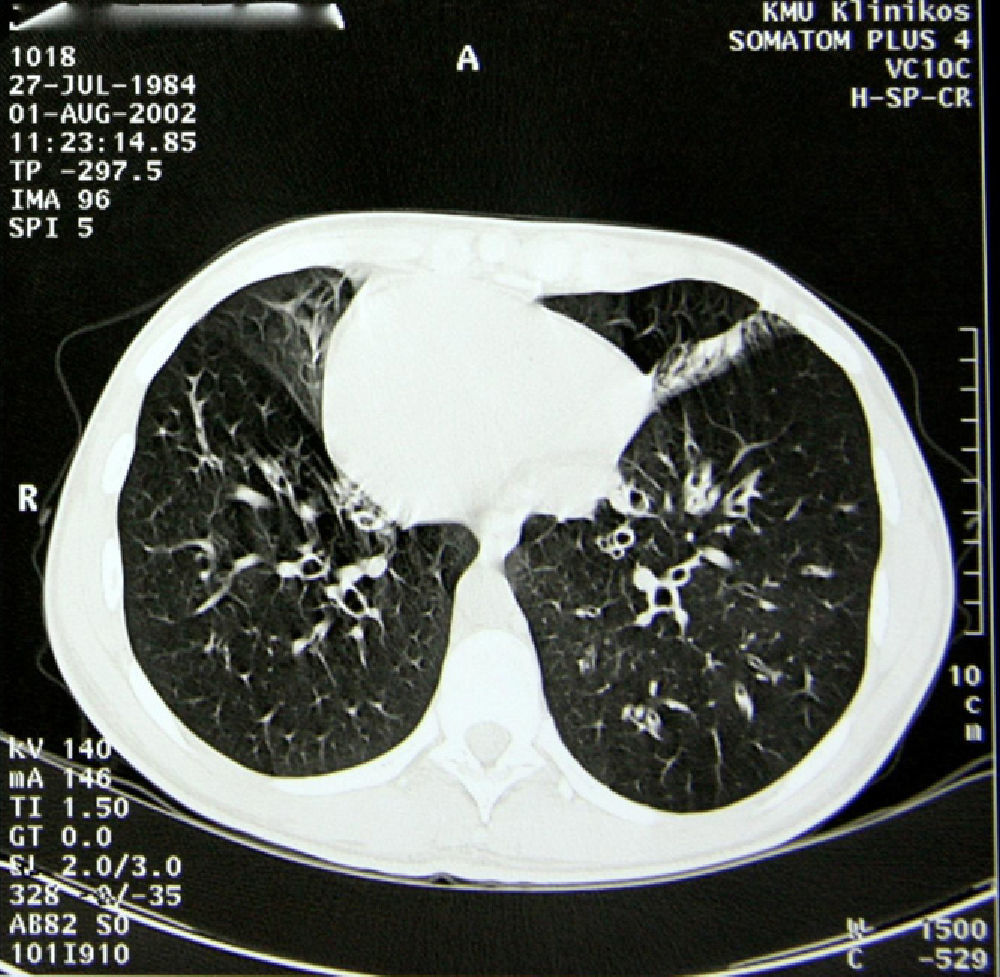
For the treatment of the bronchial obstruction, long-acting inhaled bronchodilators were administered (β2 agonists [salbutamol] at a dosage of 50μg/day). The treatment of the persistent exacerbations of the bronchiectasis consisted of short 10–14 day cycles of antibiotics and mucolytics (oral bromhexine, at a dosage of 16mg 3 times a day, was effective). The mean annual rate of lower respiratory tract infections was 2.5. Daily physiotherapy and physical exercise were applied, nutrition was optimized and the patient avoided environmental contaminants (including tobacco smoke). All these methods reduced the patient's symptoms, lowered her inflammatory marker levels and improved her quality of life. Spirometry was repeated once a year. Lung function did not deteriorate during the 7-year follow-up: in order to supervise the progression of the disease, every 2 years thoracic CT was carried out. Radiological findings did not demonstrate changes and the patient had not developed new bronchiectasis (Fig. 1).
DiscussionThe clinical characteristics of Kartagener's syndrome are productive cough, respiratory tract infections, sinusitis, otitis media and infertility.7,8 In PCD, the clinical phenotype is intensive and it overlaps with other chronic diseases of the respiratory tract.8 The defect is congenital, and symptoms present at an early age, which emphasizes the importance for pediatricians to know this disease as a substantial, although infrequent, differential diagnosis in children with recurring symptoms in the upper and lower respiratory tract.5
In the patient we describe, the diagnosis of Kartagener's syndrome was established at the age of 14. Frequently, the diagnosis of PCD is delayed until adolescence or adulthood as a consequence of the heterogeneous nature of the disease, the lack of physicians’ knowledge about the characteristics of the disease and the technical experience that is necessary for a precise diagnosis.3,8 In addition, the diagnosis of PCD may be delayed because the syndrome, characterized by bronchitis, sinusitis and otitis, can be easily confused with common infections. The delayed diagnosis of the disease can translate into adverse consequences for the patient, including insufficient care or inappropriate treatment.8,9
It has been found that other genetic markers detected in these patients (AAT and IgA) are protectors (due to the high concentration). The primary function of AAT is to inhibit neutrophil elastase in the lung interstitium and the alveolar space. The increase in serum levels of AAT may reflect a beneficial change of the protease–antiprotease balance, the fundamental element of the physiopathological pathway that mediates the effect of congenital AAT deficiency in chronic obstructive pulmonary disease (COPD) and bronchiectasis. The detection of higher levels of AAT and IgA was unrelated with the inflammatory state (C-reactive protein levels were normal). We formulated the hypothesis that these factors would protect against infection and the additional progression of bronchiectasis.
The respiratory treatment of bronchiectasis includes respiratory supervision at regular intervals, respiratory tract clearance with combinations of physiotherapy and physical exercise, and an aggressive treatment of upper and lower respiratory tract infections.10 The patient we report received an ample spectrum of curative methods: short cycles of antibiotics, mucolytics, long-acting bronchodilators and daily physical therapy. In general, antibiotics are used during exacerbations of the disease and they are prescribed according to the bacterial growth from the previous sputum culture. The objective of the treatment should be the prevention of chronic lung lesions and bronchiectasis.11 The two pillars of respiratory treatment are antibiotic therapy and thoracic physiotherapy. Physiotherapy is essential to improve the clearance of the respiratory tract with the aim to delay the onset and progression of obstructive respiratory disease.4,10 Physical exercise can contribute to sputum clearance, and it has been demonstrated to be a better bronchodilator than the use of bronchodilators themselves in PCD.9 The prognosis is generally considered favorable, and life expectancy is usually normal. An important part of the clinical visits at regular intervals should be monitoring the progression of the lung disease.3 The described patient was followed up with regularly scheduled appointments every 6 months, including additional visits during the exacerbations.
The present clinical case demonstrated a non-progressive course of the bronchiectasis, even given the congenital Kartagener's syndrome, which therefore indicates that the progression of bronchiectasis is a complex interrelationship between genetic variation and an appropriate non-specific treatment.
Please cite this article as: Serapinas D, et al. Una regresión poco común de los síntomas de un síndrome de Kartagener. Arch Bronconeumol. 2012;49:28–30.







