
Tras la presentación de un caso de enfermedad de Niemann-Pick (N-P) tipo B se realiza una revisión de la literatura en la que se describen los hallazgos radiológicos, con especial hincapié en la afectación pulmonar. Se trata de un paciente de 40 años diagnosticado a los 6 años de posible enfermedad de N-P sospechada por presentar hepatoesplenomegalia y patrón reticular en radiografía de tórax, con pruebas de función respiratoria inicialmente normales. Dicho diagnóstico se confirmó mediante medición de la actividad de la enzima esfingomielinasa ácida (ASM) en cultivo de fibroblastos de piel y demostración de la presencia de mutaciones en el gen de la ASM. Ante una enfermedad de depósito habría que tener siempre en cuenta la afectación pulmonar, y la técnica radiológica más fiable es la tomografía computarizada de alta resolución. Aunque el patrón encrazy-paving no es el más específico de esta patología, la enfermedad de N-P debería estar en el diagnóstico diferencial.
We have carried out a review of the literature after the presentation of a case of Niemann-Pick (N-P) disease type B reporting the radiological findings with special emphasis on lung affectation. The patient is 40 years old and was diagnosed at the age of 6 with possible N-P disease suspected due to the presentation of hepatosplenomegaly and reticular pattern on chest radiography, with initially normal lung function tests. Said diagnosis was confirmed by measuring the activity of acid sphingomyelinase (ASM) enzyme in skin fibroblast cultures and by demonstrating the presence of mutations in the ASM gene. In cases of storage disease, lung affectation should always be considered, and the most reliable radiological technique is high-resolution computed tomography. Although the “crazy-paving” pattern is not the most specific for this pathology, N-P disease should be included in the differential diagnosis.
La enfermedad de Niemann-Pick (N-P) es un trastorno de almacenamiento lisosómico en el que se produce acumulación de esfingomielina1 en diferentes tejidos. Da lugar a una afectación multiorgánica y clásicamente se ha dividido en dos subtipos de acuerdo a su fenotipo: tipos A y B1. Se describe un tercer subtipo, el tipo C, que está producido por una alteración genética diferente. En el tipo A se produce un trastorno neurodegenerativo con muerte prematura. El tipo B produce una afectación crónica visceral2, afectándose de forma tardía y con mucha frecuencia el pulmón. Radiográficamente la afectación pulmonar viene caracterizada por un patrón intersticial asociado a opacidades en vidrio deslustrado. Existe una débil correlación entre la función pulmonar y la enfermedad intersticial3. Se presenta un caso de enfermedad de N-P tipo B con afectación pulmonar y hepática. El objetivo es exponer los hallazgos patológicos de esta enfermedad en el pulmón tanto en la radiografía simple de tórax como en la tomografía computarizada de alta resolución (TACAR), su correlación con los test de función pulmonar y revisar la literatura.
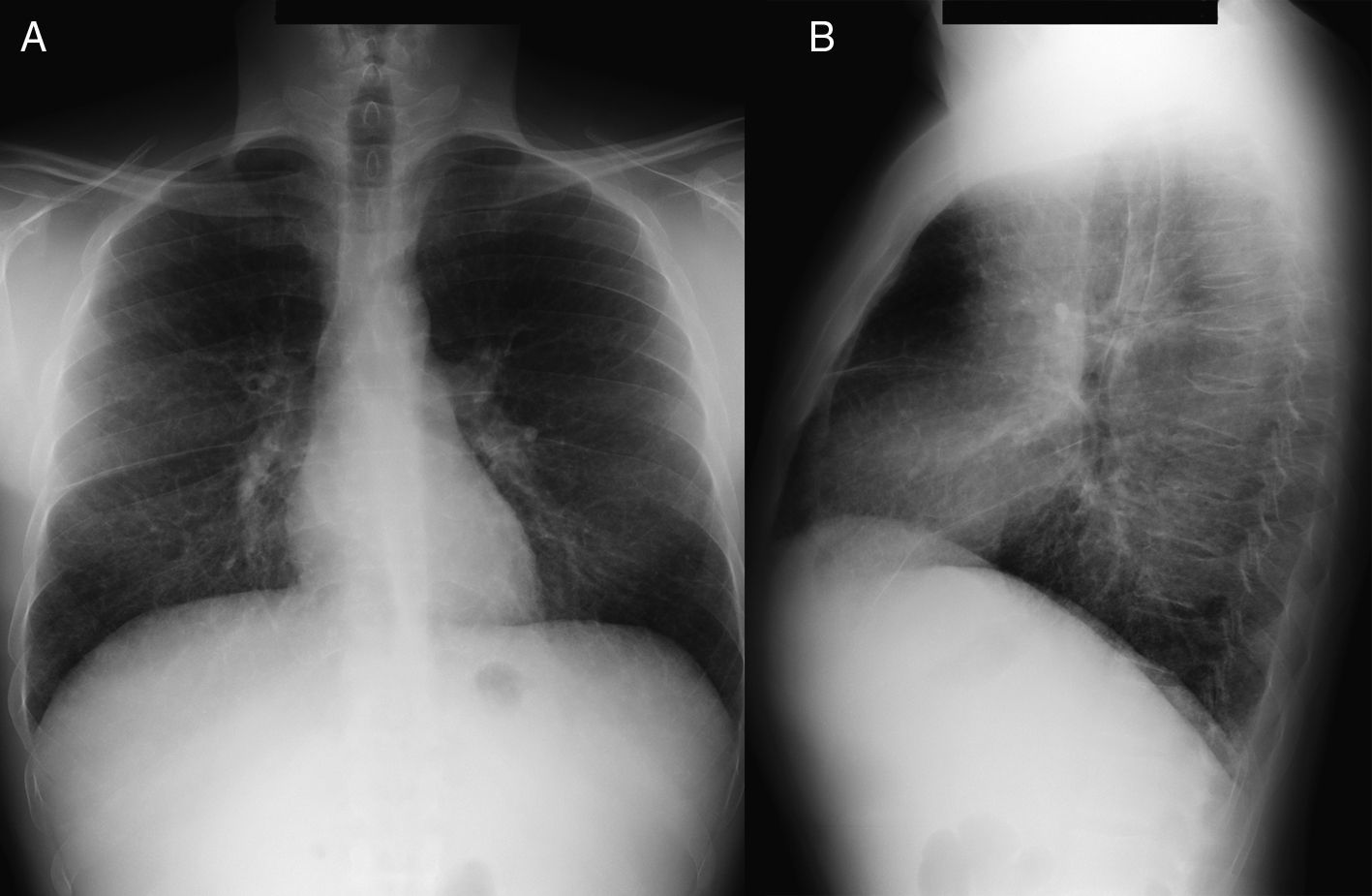
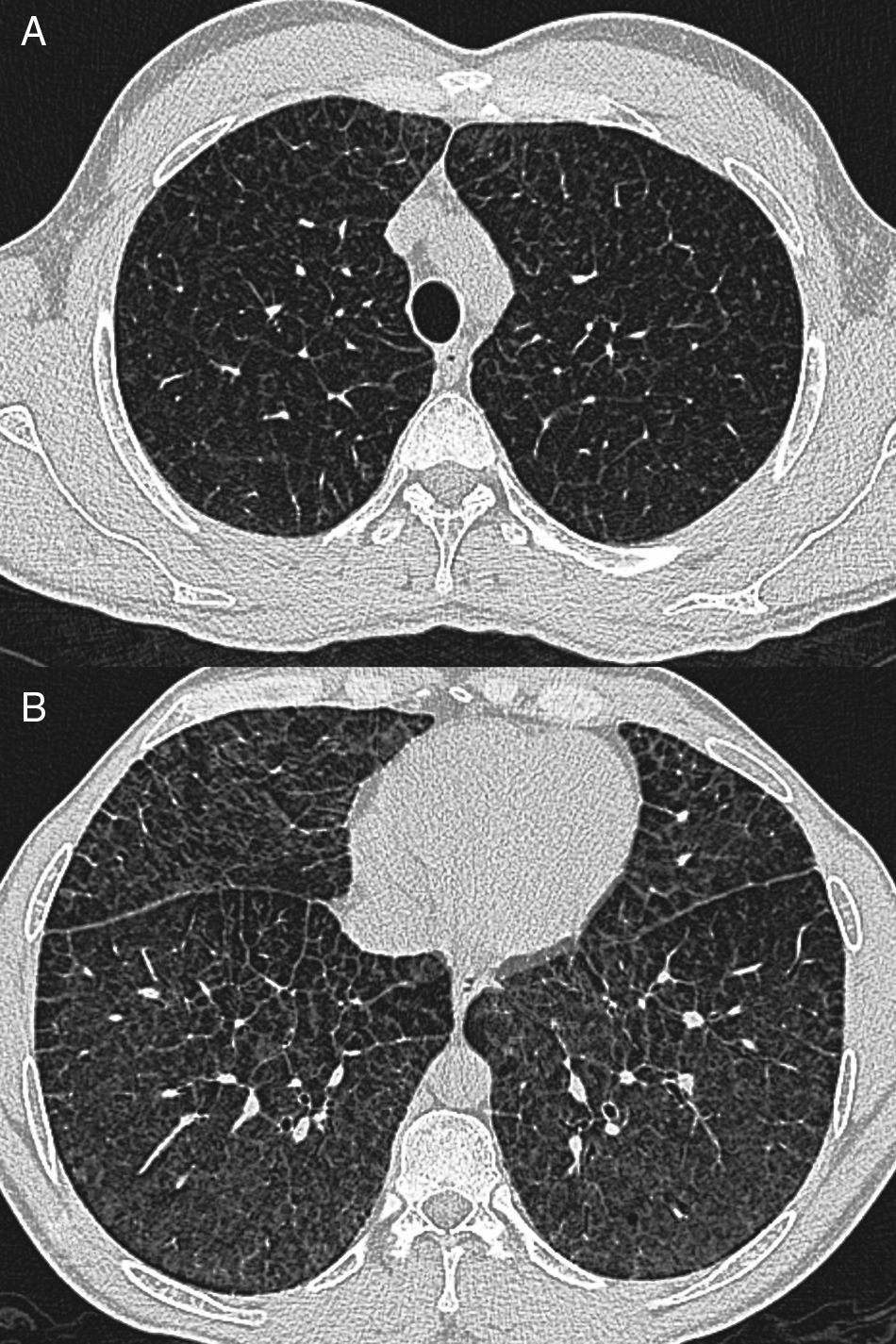
Caso clínicoPaciente varón de 40 años, diagnosticado de enfermedad de N-P con afectación hepática y pulmonar. Antecedentes personales: hepatoesplenomegalia diagnosticada desde los 6-7 meses de vida. Ingresa a los 6 años de edad por cuadro de astenia, anorexia, decaimiento y fiebre. Se le realizó radiografía de tórax posteroanterior y lateral (PA-L), en la que se apreció un patrón reticular difuso. Ante los hallazgos radiográficos y la presencia de hepatoesplenomegalia se sospechó enfermedad por depósito, realizándose biopsia de la mucosa rectal y biopsia hepática, que fueron sugestivas de enfermedad de N-P (se visualizaron macrófagos cargados de lípidos). La exploración neurológica y las pruebas de función respiratoria fueron normales. Se confirmó el diagnóstico a los 20 años mediante biopsia de piel con cultivo de fibroblastos, comprobándose un marcado déficit de actividad de esfingomielinasa ácida (ASM) característica de enfermedad N-P que, apoyando el diagnóstico clínico, sería tipo B. A los 38 años se realizó el diagnóstico molecular, en el que se observaron dos mutaciones en el gen SMPD (presentó heterocigosis en mutación p.R608del). Actualmente sigue revisiones en consultas de neumología y en consultas de hepatología, y presenta ictericia intermitente y disnea de máximos esfuerzos (grado 0 MRC). En la exploración física destaca la presencia de mínimos crepitantes bibasales, ictericia subconjuntival y hepatoesplenomegalia. La exploración neurológica es normal. En la analítica los resultados más destacables son: bilirrubina total, 4,17mg/dl; GOT, 38UI/l; GPT, 45UI/l; colesterol total, 221mg/dl. Las pruebas de función respiratoria muestran un patrón ventilatorio normal (FVC,3,84 [87,7%]; FEV1, 3,13 [86%]; FEV1/FVC, 81,58%; TLC, 5,26 [81,8%]) y una alteración leve del intercambio gaseoso (DLCO, 57,5%). Hallazgos radiográficos: radiografía PA-L de tórax (fig. 1 A-B): en el parénquima pulmonar se observa una afectación intersticial reticular difusa de predominio en campos medios e inferiores; TCAR (Aquilion Toshiba de 64 detectores) (fig. 2 A y B): afectación en vidrio deslustrado difusa aunque de predominio en los lóbulos inferiores con engrosamientos septales subpleurales en la pleura periférica, mediastínica y cisuras. No se observan adenopatías mediastínicas ni hiliares. El paciente actualmente no sigue ningún tratamiento y únicamente presenta afectación hepática y pulmonar sin que haya afectación del sistema nervioso central.
La enfermedad de N-P fue descrita por Albert Niemann en 19144. Se trata de un trastorno de almacenamiento lisosómico1 autosómico recesivo. Su incidencia es de 0,4 a 0,6/100.000 recién nacidos3. Es el resultado de la deficiencia de ASM y la acumulación de esfingomielina1 principalmente en el sistema nervioso central y el sistema reticuloendotelial5 en forma de macrófagos cargados de lípidos llamados «células de Niemann-Pick»6. La enfermedad de N-P engloba dos entidades diferentes en términos de metabolismo y síntomas clínicos. Tipo A y B: deficiencia heredada de la actividad de ASM; se caracterizan por la acumulación de lípidos en diferentes tejidos, sobre todo en el hígado y el bazo6. Tipo C: trastorno de almacenamiento lipídico de mayor complejidad debido al defecto del tráfico de colesterol intracelular6, producido por alteración genética diferente.
El tipo A es una enfermedad neurodegenerativa severa de la infancia con retraso psicomotor progresivo, fallo para medrar, hepatoesplenomegalia y máculas rojo fresa2. La esperanza de vida es corta, 3-4 años para la mayoría4.
El tipo B no parece tener predilección por el sexo y se manifiesta a cualquier edad, aunque es más frecuente antes de los 20 años7. Existe una afectación crónica visceral que se desarrolla de forma lenta2 y presenta una amplia variabilidad en la severidad de los síntomas y los hallazgos clínicos3. La hepatoesplenomegalia es bastante común, con crecimiento esplénico sobre todo1. Se afectan las pruebas de función hepática con aumento de transaminasas y bilirrubina. Puede aparecer trombocitopenia, que podría ser secundaria a infiltración de la médula ósea por células de N-P8, y también puede producir leucopenia. La enfermedad pulmonar es frecuente, y en la mayoría de pacientes aparecen alteraciones en la radiografía o la TACAR de tórax1. La incidencia exacta de la afectación pulmonar es difícil de determinar, pero es raro en adultos y más común en las formas infantiles9. Las células de N-P se acumulan en los septos alveolares, en las paredes bronquiales y en pleura y ocasionan un patrón restrictivo que empeora progresivamente, pudiendo llegar a fallo respiratorio1 con dependencia de oxígeno y/o reducción de la tolerancia al ejercicio físico10. Las pruebas de función respiratoria muestran dicho patrón restrictivo con incapacidad para el intercambio gaseoso, y la ergometría cíclica es anormal en la mitad de los pacientes3. Los hallazgos radiológicos vienen determinados por la existencia de un patrón reticulonodular con engrosamiento de septos interlobulares, opacidades en vidrio deslustrado y a veces nódulos subcentimétricos que pueden estar calcificados2,8,10. En ocasiones la combinación de estos hallazgos produce la imagen llamada crazy paving en TACAR, si bien estos hallazgos no son específicos. El diagnóstico diferencial de este patrón incluye: edema pulmonar, hemorragia pulmonar, linfangitis, proteinosis alveolar, amiloidosis3 y neumonía intersticial no específica. Sin embargo, la asociación con hepatoesplenomegalia debería sugerir enfermedad por depósito2. Existe una pobre correlación entre los test de función pulmonar y los hallazgos morfológicos en la TACAR, lo que implica que los cambios intersticiales no siempre afectan al intercambio gaseoso en un grado significativo3,10. Por lo tanto, la presencia de enfermedad pulmonar intersticial en la radiografía simple o en la TACAR no es necesariamente un indicador fiable de aparición de síntomas clínicos ni de la alteración de las pruebas funcionales respiratorias. Para el estudio de las alteraciones pulmonares en esta enfermedad deben emplearse tanto la radiografía simple de tórax como la TACAR1. En nuestro caso, la afectación pulmonar se evidenció a los 6 años mediante la detección de patrón reticular en la radiografía simple de tórax, pero el paciente no ha presentado hasta el momento actual clínica respiratoria significativa.
El diagnóstico se realiza por la identificación de células de N-P en aspirado de médula ósea11 o bien mediante la demostración de la reducción de la actividad de la ASM en leucocitos aislados y/o cultivo de fibroblastos de la piel. El diagnóstico molecular viene dado por la identificación de dos mutaciones de la ASM causantes de la enfermedad10.
No existe actualmente un tratamiento específico disponible para la deficiencia de la ASM. La terapia de sustitución enzimática y la terapia génica podrían ser útiles para pacientes con enfermedad tipo B3,4.
Nuestro caso presenta la mayoría de los hallazgos pulmonares radiológicos descritos en la literatura con presencia de crazy paving con gradiente ápico-basal. Las pruebas de función respiratoria que presenta no se correlacionan con las alteraciones morfológicas en la TCAR, puesto que los test respiratorios son prácticamente normales. Por tanto, creemos que aunque la enfermedad de N-P es una entidad rara que presenta unas alteraciones radiológicas inespecíficas, éstas pueden apoyar el diagnóstico inicial y su confirmación. Ante un trastorno de almacenamiento de lípidos o una enfermedad de depósito habría que tener siempre en cuenta la afectación pulmonar, y la técnica radiológica más fiable es la TACAR5. Aunque el patrón en crazy paving no es el más específico para este tipo de enfermedad, esta debería estar incluida en su diagnóstico diferencial de acuerdo con lo expresado anteriormente en la literatura5,10.







