






Most patients with idiopathic pulmonary fibrosis (IPF) treated with antifibrotics (AF) have progressive disease despite treatment. A switch of AF may improve survival, but evidence from randomised controlled trials is missing. We aimed to evaluate the efficacy of an AF switch on survival and FVC decline in patients from the European MultiPartner IPF registry (EMPIRE).
MethodsThe study included 612 patients who discontinued the first antifibrotic therapy. Patients were grouped and analysed from two perspectives: (1) whether they had received a second antifibrotic treatment after the discontinuation of the first therapy, and (2) a reason for discontinuation of the first AF – “lack of efficacy” (LE) and “intolerance” (INT).
ResultsWhile 263 (43%) of 612 patients received no second AF (“non-switched”), 349 (57%) patients switched. Overall survival was higher in patients who received a second AF (median 50 vs. 29 months; adjusted HR 0.64, P=0.023). Similarly, the annual FVC decline was significantly reduced in switched patients: −98ml/y in switched and −172ml/y in non-switched patients (P=0.023), respectively. The switched patients had similar risk for mortality in both LE and INT groups (adjusted HR 0.95, P=0.85). The high impact of switching on survival was demonstrated in LE patients (adjusted HR 0.27, P<0.001).
ConclusionThe patients without a second AF had significantly shorter overall survival. Our analysis suggests the importance of switching patients with an ineffective first AF therapy to a second AF therapy.

Idiopathic pulmonary fibrosis (IPF) is a chronic progressive lung disease characterised by loss of pulmonary function and, as reported in the pre-antifibrotic era, an estimated survival of 3–5 years from diagnosis.1 The introduction of antifibrotic (AF) treatment has led to significant improvements in IPF patients’ outcomes.2 The widely available AF medications – nintedanib and pirfenidone – exert their activity by different mechanisms. Nintedanib is a tyrosine kinase inhibitor targeting receptors for vascular endothelial growth factor, platelet-derived growth factor and others.3 The mechanism of action of pirfenidone is not fully elucidated, but downregulation of the production of procollagen and growth factors has been observed.4
The efficacy of nintedanib and pirfenidone has been clearly established in randomised clinical trials.5,6 Both can slow the progression of IPF but do not reverse lung damage. Real-world evidence of the efficacy of these treatments in terms of longer survival has been shown in recent studies; some of them demonstrated median or mean survival of patients treated with AF higher than five years7–9 and the reduction of risk of death between 25.1% and 41% in comparison to non-AF treatment.10–12
In patients who discontinue the first AF treatment, most often because of adverse events or disease progression, the availability of a second AF involving a different mechanism of action is potentially an attractive option. Small-scale studies and other reports have shown that switching to a second AF after stopping the first AF is feasible, resulting in a slower decline of lung function and longer overall survival in comparison with patients without a second AF.13–20 However, conclusive data on the effectiveness of switching from one antifibrotic to another have not yet been available.
The European MultiPartner IPF registry (EMPIRE) has continuously enrolled patients in 11 Central and Eastern European countries since 2012, resulting in a longitudinal database of well-characterized patients. We herein report the effects of switching antifibrotic therapy on FVC and survival of IPF patients included in the EMPIRE registry, focusing on the reason for the first AF treatment discontinuation.
MethodsThis analysis includes IPF patients with their first visit to the EMPIRE registry from 1 December 2014, followed through 30 September 2022. IPF was diagnosed according to the 2011 ATS/ERS/JRS/ALAT criteria.21 Final IPF diagnosis was always determined by a local multidisciplinary team; the EMPIRE data, therefore, also includes patients with HRCT or histopathological findings not demonstrating UIP pattern.
Patients in the EMPIRE registry were eligible for this study if they received either nintedanib or pirfenidone, discontinued treatment for any reason and were known to have survived for at least six months after discontinuation. Patients who died within six months of discontinuation were excluded from the study as they did not have time to start a second antifibrotic therapy and would be over-represented in the non-switched group, resulting in immortal time bias.
The patients were grouped and analysed from two perspectives: (1) whether they had received a second antifibrotic treatment after the discontinuation of the first therapy (hereinafter referred to as switch and non-switch group), and (2) a reason for discontinuation of the first AF – “lack of efficacy” (LE) and “intolerance” (INT). The LE group included patients who experienced a significant IPF progression, and the first AF treatment was considered ineffective according to the treating physician's opinion or, in some countries, by rules for the innovative therapy administration and termination (so-called “stopping rules”). The INT group included patients who discontinued the first AF treatment due to adverse events, non-compliance, refusal, financial coverage, administrative reasons, other health conditions, medical procedures etc. In other words, although the first AF treatment was effective (in terms of stable disease or slower lung deterioration), it was discontinued for a “comfort-related” reason.
Continuous parameters were described by the median and interquartile range (IQR); categorical parameters were described by absolute and relative frequencies. Differences between groups of patients were tested by Kruskal–Wallis test or Wilcoxon test for continuous parameters and by the maximum likelihood chi-square test for categorical parameters.
Kaplan–Meier methodology was used to visualising overall survival (OS) and progression-free survival (PFS) from the date of discontinuation of the first antifibrotic therapy. For analysis of PFS, progression was defined as death, lung transplantation or an absolute decline in forced vital capacity (FVC) of >10% or in diffusing capacity of the lungs for carbon monoxide of >15% from baseline. Risk factors for OS and PFS were analysed by Cox proportional hazard model and presented as hazard ratio (HR), both crude and adjusted for baseline values (sex, age and baseline FVC level).
The annual rate of change of FVC in the 24 months from baseline was analysed by a restricted maximum likelihood estimation based on a random slope and intercept model. The annual rate was described by the point estimate of the time-associated slope (including 95% confidence intervals). The univariate model was followed by the multivariate one in which the annual rates were adjusted for sex, age, and FVC at baseline. The model included fixed effects for time, baseline FVC, sex, age, as well as treatment-by-time interaction. Random effects for time and intercept were included for each patient. The model was also used to test the statistical significance of the difference in slope between groups after discontinuing the first antifibrotic (no further antifibrotic, nintedanib after discontinuation of pirfenidone, pirfenidone after discontinuation of nintedanib). Statistical analyses were carried out using R software. The level of significance α was set at 0.05. Any P-values presented are considered nominal in nature, and no adjustment for multiplicity was made.
Other parameters analysed at baseline only were the GAP index22 and dyspnoea rated according to the NYHA criteria.23
ResultsPatients’ CharacteristicsA total of 1270 patients were identified in the EMPIRE registry between December 2014 and September 2022 who had terminated their first antifibrotic therapy. Of these, 612 patients were followed up after the termination of the first antifibrotic therapy for at least six months and were included in the analysis. The patients were from Czechia (290), Turkey (120), Poland (58), Israel (51), Hungary (39), Austria (26), Croatia (9), Serbia (9) and Bulgaria (2). Of those included, 263 (43%) did not receive a second antifibrotic: the first AF therapy was pirfenidone in 164 (27%) patients and nintedanib in 99 (16%) patients, while 349 (57%) patients switched; namely, 206 (34%) from pirfenidone to nintedanib and 143 (23%) from nintedanib to pirfenidone. The median follow-up of the patients was 20 months. The remaining 658 patients who discontinued their first AF treatment were not followed up further due to death or loss to follow-up within the first 6 months of discontinuation.
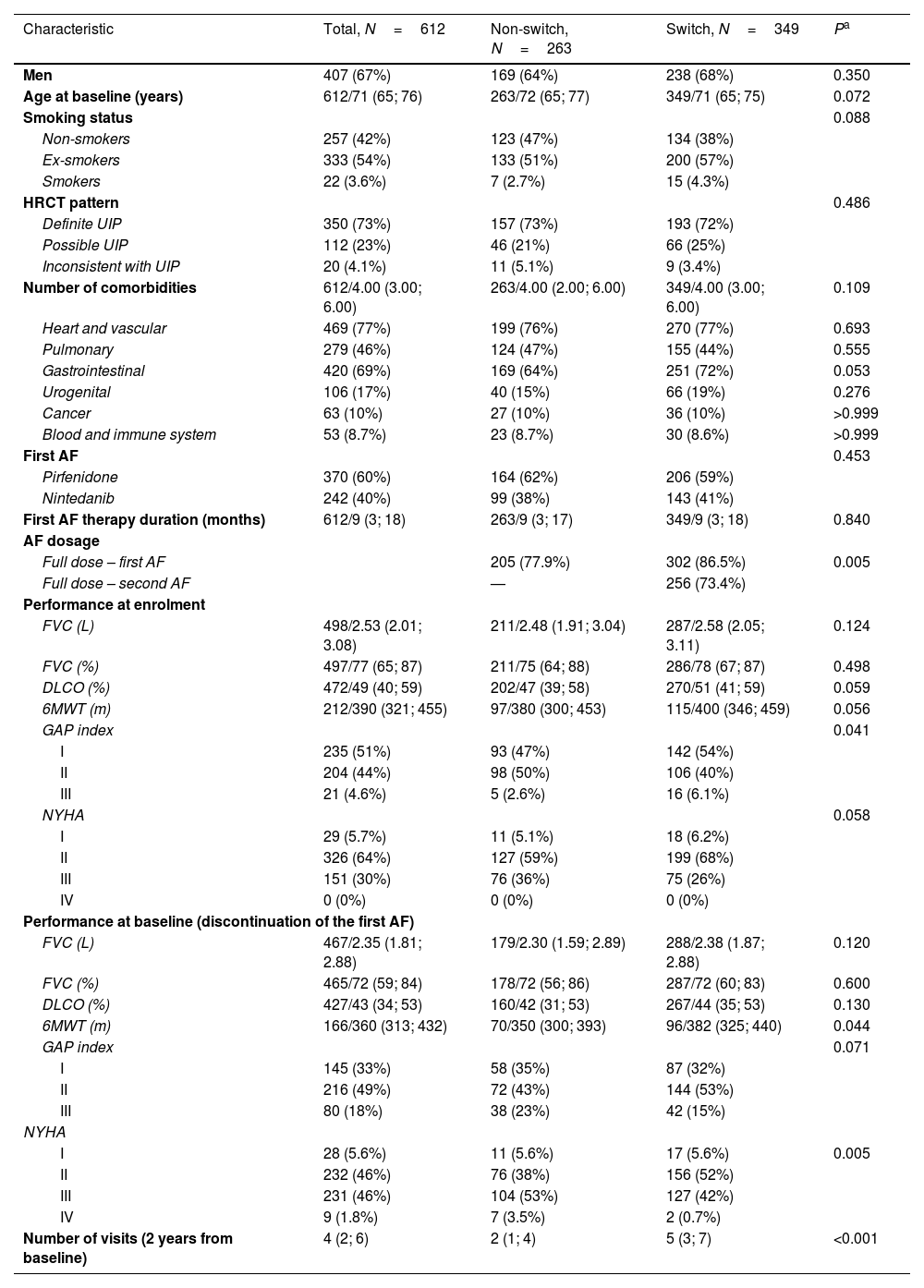
Baseline characteristics of the switched and non-switched patients and P-values for significant differences are shown in Table 1. There was a somewhat higher proportion of men and a lower proportion of non-smokers among the switched patients. Patients in this group were also slightly younger and had better performance at baseline – better values of pulmonary function tests and a higher proportion of GAP I and NYHA I+II stages.
Characteristics of Patients Who Discontinued Their First Antifibrotic Therapy.
| Characteristic | Total, N=612 | Non-switch, N=263 | Switch, N=349 | Pa |
|---|---|---|---|---|
| Men | 407 (67%) | 169 (64%) | 238 (68%) | 0.350 |
| Age at baseline (years) | 612/71 (65; 76) | 263/72 (65; 77) | 349/71 (65; 75) | 0.072 |
| Smoking status | 0.088 | |||
| Non-smokers | 257 (42%) | 123 (47%) | 134 (38%) | |
| Ex-smokers | 333 (54%) | 133 (51%) | 200 (57%) | |
| Smokers | 22 (3.6%) | 7 (2.7%) | 15 (4.3%) | |
| HRCT pattern | 0.486 | |||
| Definite UIP | 350 (73%) | 157 (73%) | 193 (72%) | |
| Possible UIP | 112 (23%) | 46 (21%) | 66 (25%) | |
| Inconsistent with UIP | 20 (4.1%) | 11 (5.1%) | 9 (3.4%) | |
| Number of comorbidities | 612/4.00 (3.00; 6.00) | 263/4.00 (2.00; 6.00) | 349/4.00 (3.00; 6.00) | 0.109 |
| Heart and vascular | 469 (77%) | 199 (76%) | 270 (77%) | 0.693 |
| Pulmonary | 279 (46%) | 124 (47%) | 155 (44%) | 0.555 |
| Gastrointestinal | 420 (69%) | 169 (64%) | 251 (72%) | 0.053 |
| Urogenital | 106 (17%) | 40 (15%) | 66 (19%) | 0.276 |
| Cancer | 63 (10%) | 27 (10%) | 36 (10%) | >0.999 |
| Blood and immune system | 53 (8.7%) | 23 (8.7%) | 30 (8.6%) | >0.999 |
| First AF | 0.453 | |||
| Pirfenidone | 370 (60%) | 164 (62%) | 206 (59%) | |
| Nintedanib | 242 (40%) | 99 (38%) | 143 (41%) | |
| First AF therapy duration (months) | 612/9 (3; 18) | 263/9 (3; 17) | 349/9 (3; 18) | 0.840 |
| AF dosage | ||||
| Full dose – first AF | 205 (77.9%) | 302 (86.5%) | 0.005 | |
| Full dose – second AF | — | 256 (73.4%) | ||
| Performance at enrolment | ||||
| FVC (L) | 498/2.53 (2.01; 3.08) | 211/2.48 (1.91; 3.04) | 287/2.58 (2.05; 3.11) | 0.124 |
| FVC (%) | 497/77 (65; 87) | 211/75 (64; 88) | 286/78 (67; 87) | 0.498 |
| DLCO (%) | 472/49 (40; 59) | 202/47 (39; 58) | 270/51 (41; 59) | 0.059 |
| 6MWT (m) | 212/390 (321; 455) | 97/380 (300; 453) | 115/400 (346; 459) | 0.056 |
| GAP index | 0.041 | |||
| I | 235 (51%) | 93 (47%) | 142 (54%) | |
| II | 204 (44%) | 98 (50%) | 106 (40%) | |
| III | 21 (4.6%) | 5 (2.6%) | 16 (6.1%) | |
| NYHA | 0.058 | |||
| I | 29 (5.7%) | 11 (5.1%) | 18 (6.2%) | |
| II | 326 (64%) | 127 (59%) | 199 (68%) | |
| III | 151 (30%) | 76 (36%) | 75 (26%) | |
| IV | 0 (0%) | 0 (0%) | 0 (0%) | |
| Performance at baseline (discontinuation of the first AF) | ||||
| FVC (L) | 467/2.35 (1.81; 2.88) | 179/2.30 (1.59; 2.89) | 288/2.38 (1.87; 2.88) | 0.120 |
| FVC (%) | 465/72 (59; 84) | 178/72 (56; 86) | 287/72 (60; 83) | 0.600 |
| DLCO (%) | 427/43 (34; 53) | 160/42 (31; 53) | 267/44 (35; 53) | 0.130 |
| 6MWT (m) | 166/360 (313; 432) | 70/350 (300; 393) | 96/382 (325; 440) | 0.044 |
| GAP index | 0.071 | |||
| I | 145 (33%) | 58 (35%) | 87 (32%) | |
| II | 216 (49%) | 72 (43%) | 144 (53%) | |
| III | 80 (18%) | 38 (23%) | 42 (15%) | |
| NYHA | ||||
| I | 28 (5.6%) | 11 (5.6%) | 17 (5.6%) | 0.005 |
| II | 232 (46%) | 76 (38%) | 156 (52%) | |
| III | 231 (46%) | 104 (53%) | 127 (42%) | |
| IV | 9 (1.8%) | 7 (3.5%) | 2 (0.7%) | |
| Number of visits (2 years from baseline) | 4 (2; 6) | 2 (1; 4) | 5 (3; 7) | <0.001 |
Data presented as no. of patients (%) OR no. of patients/median value (25th percentile; 75th percentile).
The most frequent reason for discontinuation of the first antifibrotic therapy were side effects (50% overall, 45% among those discontinuing pirfenidone and 57% among those discontinuing nintedanib), followed by lack of efficacy/stopping rules (18% overall, 19% of those discontinuing pirfenidone and 16% of those discontinuing nintedanib) (Table 2).
Reasons for Discontinuing the First Antifibrotic Therapy.
| Characteristic | Total, N=612 | Non-switch PIR, N=164 | Non-switch NIN, N=99 | Switch PIR–NIN, N=206 | Switch NIN–PIR, N=143 |
|---|---|---|---|---|---|
| Lack of efficacy | 108 (18%) | 26 (16%) | 15 (15%) | 43 (21%) | 24 (17%) |
| Adverse events | 304 (50%) | 51 (31%) | 54 (55%) | 116 (56%) | 83 (58%) |
| Refusal, non-compliance | 93 (15%) | 44 (27%) | 14 (14%) | 25 (12%) | 10 (7.0%) |
| Financial, administrative, health-related and other | 107 (17%) | 43 (26%) | 16 (16%) | 22 (11%) | 26 (18%) |
Data presented as no. of patients (%).
PIR, pirfenidone; NIN, nintedanib.
The overall survival (OS) rates were significantly higher in patients who received a second antifibrotic than in those who did not (median 50 vs. 29 months, HR 0.54, 95% CI: 0.42–0.7, P<0.001). OS rates at three years were 61% in patients receiving a second antifibrotic and 42% in those not receiving a second antifibrotic; HR adjusted for sex, age and FVC at baseline was 0.62 (95% CI: 0.46–0.86, P=0.004) (Fig. 1).

When we stratified patients according to their first AF, a similar effect of switching was observed. Patients who discontinued pirfenidone reached higher OS rates if switched to nintedanib (median 50 vs. 29 months, HR 0.54, 95% CI: 0.4–0.75, P<0.001), with adjusted mortality HR 0.64 (95% CI: 0.43–0.94, P=0.023), compared to non-switchers. Similarly, patients switched from nintedanib to pirfenidone had higher survival rates than those not switched to a second AF (median not reached vs. 34 months, P=0.023, HR 0.55, 95% CI: 0.34–0.89, P=0.015), with adjusted mortality HR 0.59 (95% CI: 0.33–1.07, P=0.083). See Supplementary Table 1 and Supplementary Figure 1 for details.
The progression-free survival (PFS) rates were comparable in the switched and non-switched group – the median was 14 months in patients who received a second antifibrotic and 17 months in those who did not (P=0.180), with adjusted HR for progression 1.23 (95% CI: 0.98–1.54, P=0.076) (Fig. 2).

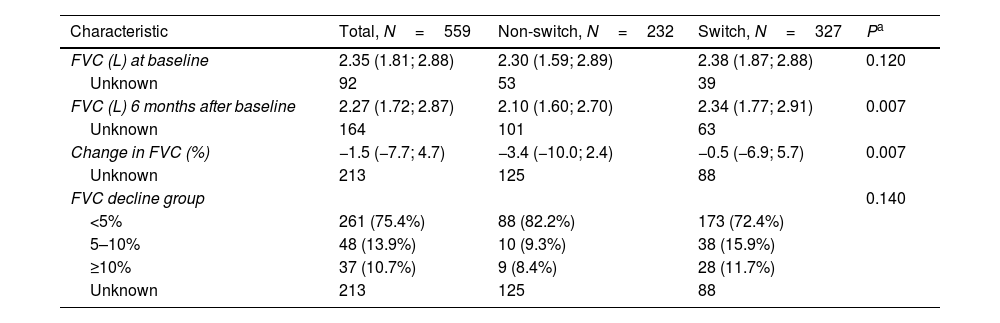
The FVC decline was 0.2–3.9% (calculated from absolute FVC values) between baseline (i.e., the date of termination of the first antifibrotic therapy) and six months post-baseline. In 346 patients with FVC measurements at both time points, there were significant differences in absolute FVC decline between switched and non-switched patients (the median decline of 0.5% vs. 3.4%, P=0.007, Table 3).
The Annual Rate of FVC Decline, Adjusted for Age, Sex and Baseline FVC.
| Characteristic | Total, N=559 | Non-switch, N=232 | Switch, N=327 | Pa |
|---|---|---|---|---|
| FVC (L) at baseline | 2.35 (1.81; 2.88) | 2.30 (1.59; 2.89) | 2.38 (1.87; 2.88) | 0.120 |
| Unknown | 92 | 53 | 39 | |
| FVC (L) 6 months after baseline | 2.27 (1.72; 2.87) | 2.10 (1.60; 2.70) | 2.34 (1.77; 2.91) | 0.007 |
| Unknown | 164 | 101 | 63 | |
| Change in FVC (%) | −1.5 (−7.7; 4.7) | −3.4 (−10.0; 2.4) | −0.5 (−6.9; 5.7) | 0.007 |
| Unknown | 213 | 125 | 88 | |
| FVC decline group | 0.140 | |||
| <5% | 261 (75.4%) | 88 (82.2%) | 173 (72.4%) | |
| 5–10% | 48 (13.9%) | 10 (9.3%) | 38 (15.9%) | |
| ≥10% | 37 (10.7%) | 9 (8.4%) | 28 (11.7%) | |
| Unknown | 213 | 125 | 88 |
Data presented as no. of patients (%) OR no. of patients/median value (25th percentile; 75th percentile).
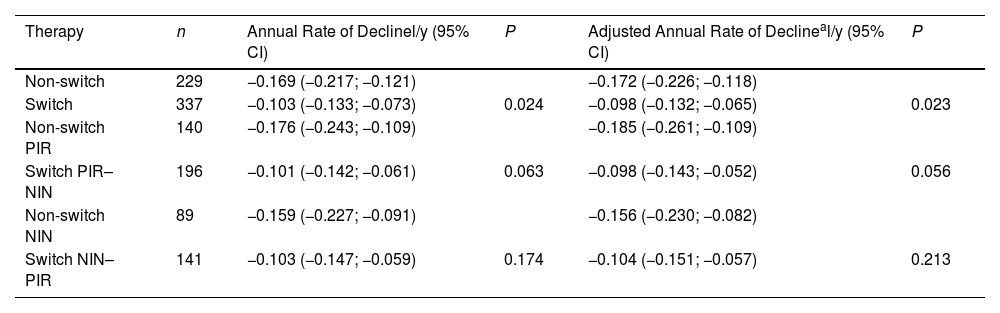
The annual FVC decline rates calculated by a linear model were also associated with receiving a second antifibrotic: the adjusted values were −0.098l/y (95% CI −0.132 to −0.065l/y) in switched patients and −0.172l/y (95% CI −0.226 to −0.118l/y) in non-switched patients, P=0.023. After the stratification according to the type of the first drug, the differences were still perceptible but below the level of statistical significance (Table 4).
Linear Model for Change in FVC (L).
| Therapy | n | Annual Rate of Declinel/y (95% CI) | P | Adjusted Annual Rate of Declineal/y (95% CI) | P |
|---|---|---|---|---|---|
| Non-switch | 229 | −0.169 (−0.217; −0.121) | −0.172 (−0.226; −0.118) | ||
| Switch | 337 | −0.103 (−0.133; −0.073) | 0.024 | −0.098 (−0.132; −0.065) | 0.023 |
| Non-switch PIR | 140 | −0.176 (−0.243; −0.109) | −0.185 (−0.261; −0.109) | ||
| Switch PIR–NIN | 196 | −0.101 (−0.142; −0.061) | 0.063 | −0.098 (−0.143; −0.052) | 0.056 |
| Non-switch NIN | 89 | −0.159 (−0.227; −0.091) | −0.156 (−0.230; −0.082) | ||
| Switch NIN–PIR | 141 | −0.103 (−0.147; −0.059) | 0.174 | −0.104 (−0.151; −0.057) | 0.213 |
PIR, pirfenidone; NIN, nintedanib.
We split the analysed cohort into two groups according to the reason for which their first AF therapy was terminated (stated by the attending physician): (1) the “lack of efficacy” group (LE) – fast IPF progression; (2) the “intolerance” group (INT) – adverse events, non-compliance, patient's refusal, and other reasons. The aim was to examine whether switching has a different effect in these groups and to identify patients who can profit from switching.
The baseline characteristics of these groups are given in Supplementary Tables 2 to 4. Assignment of patients to the LE or INT group was confirmed by the calculation of FVC decline during the first AF therapy: adjusted annual rates of FVC decline were −0.187l/y in the LE group and −0.075l/y in the INT group (P<0.001) (Supplementary Table 5).
The LE group had lower OS rates in comparison to the INT group in both switched (median 43 months vs. 60 months) and non-switched (median 15 months vs. 34 months) patients (Fig. 3). After adjustment, the switched patients had similar risk for mortality in both LE and INT groups (adjusted HR 0.95, 95% CI: 0.58–1.55, P=0.850) (Fig. 3A). On the other hand, in the non-switched group, the INT patients had a significantly lower risk of death than the LE patients (adjusted HR 0.43, 95% CI: 0.25–0.75, P=0.003) (Fig. 3B). The comparison of switched and non-switched patients and thus the importance of switching in patients discontinued due to the lack of efficacy of the first AF therapy is demonstrated in Fig. 4 (the median OS rates 43 vs. 15 months; adjusted HR 0.27, 95% CI 0.14–0.54, P<0.001).
 switched patients and (b) non-switched patients.")

The antifibrotics switch in IPF patients has been addressed in several studies after the second antifibrotic agent (nintedanib) was approved for the treatment. There are limited options to investigate the effect of a switch in randomised clinical trials; real-life studies and registries, often retrospective, are a reasonable way to do it, but with respect to their nature and data availability.
Our study is the largest real-life study to date investigating the effect of switching AF drugs on IPF progression and survival. We found significantly higher survival rates in switched patients than those who discontinued the AF therapy. Five-year survival rates of 30.4% and median survival of 33.7 months reported by Suzuki et al.19 were lower than in our study. Nevertheless, this difference can be partly attributed to a different methodology; their baseline was set to the second AF therapy initiation.
Previous studies evaluated the effect of switching AF therapy on a short-term basis, focusing on FVC decline, progression, or other events. The reported outcome varied, with the proportion of patients with a stabilised FVC ranging from 50% to 70%.13,17,18,24 We observed a similar proportion of patients with stable FVC after six months, even in the non-switched group; on the other hand, the decline (expressed as a percentual change of initial absolute FVC) was lower in the switched group. However, the reliability of the longitudinal analysis of FVC in real-life settings strongly depends on data (non-)availability at various time points, particularly if a decline after a specific period (6 months, 12 months) is assessed. Analysis of annual decline rates using a linear regression model may be more appropriate since it allows for including patients with PFTs outside these specific time points, and the adjustment for potential confounders is possible. We report a significant difference in adjusted annual decline rates between the switched and non-switched patients, which is consistent with findings by Adams et al.20
The progression-free survival rates were comparable in switched and non-switched patients. This may contradict the differences in FVC decline observed in these two groups, as the FVC decline is a part of the definition of disease progression. We assume that this discrepancy could be connected with the real-life registry-based nature of this study. Patients treated with antifibrotics had more frequent follow-up visits (Table 1) and, thus, a higher probability of earlier progression detection.
The previous studies were based on individuals or only a small number of patients. Our study included 349 patients who switched from one antifibrotic medication to another. Our main objective was to examine the importance of switching after the first antifibrotic therapy is discontinued, usually due to adverse events or lack of efficacy. Therefore, we did not analyse the outcomes of the first antifibrotic; we put patients on the same “starting line” after its discontinuation and focused on the benefit of the second antifibrotic therapy compared to patients who did not receive one. There might be a potential risk in this approach due to different disease courses and time from diagnosis or the first AF therapy initiation to our baseline. However, the analysis of baseline characteristics demonstrated that individual groups of patients were comparable; the aforementioned risk was low and could be overcome by usual adjustments for basic parameters (sex, age, baseline FVC). Furthermore, the size of our cohorts (switched and non-switched) was representative enough to cover differences in patient characteristics and clinical course.
There are several limitations to our study. Patients who died or were lost to follow-up within six months of discontinuing the first antifibrotic were excluded from the analysis to avoid immortal time bias. Patients were not randomised to receive antifibrotic treatment. Therefore, the choice of whether to use a second antifibrotic may have been influenced by the patient's response to the first therapy, his/her overall status and willingness to continue with further treatment or by the different approaches of the treating physicians. Considering the real-life nature of the clinical registry, the compared groups of patients also differed slightly in some parameters (sex, age, lung function). Therefore, all important conclusions from this study are based on analyses adjusted for these parameters, both for lung function assessment and HR for mortality.
Some potential agents currently being investigated in clinical trials will likely reach clinical practice.25,26 Nevertheless, pirfenidone and nintedanib, which have pharmacologically different modes of action, will be the only feasible IPF management strategy for a certain time. If a first AF treatment is discontinued, most often for adverse events and lack of efficacy (either declared by a physician or defined by reimbursement criteria), switching to the second AF is one of the few options that remain on the table. Our data confirm previous findings from small studies that indicate that switching antifibrotics is a feasible strategy for patients with IPF and further indicate that this strategy has the potential to improve long-term outcomes. Since most patients discontinued therapy due to side effects, switching AF allowed a continuation of therapy that slowed progression, highlighting an important treatment opportunity for a significant proportion of patients. For patients who tolerate the first AF, the physician has the option to either keep it (despite deteriorating lung function) or to switch treatment. However, due to reimbursement mechanisms in many EMPIRE countries and the smaller number of these patients in the cohort analysed, we have only limited data. The same applies to the comparison of the two “directions” of the switch.
In summary, the results of our study emphasise the importance of switching AF treatment to the second drug if the first drug is discontinued due to side effects or other reasons. Furthermore, our results suggest that patients with progressive disease and ineffective first AF treatment may significantly profit from the switch and have longer overall survival than patients without a second AF.
Prior PublicationATS Conference 2023 (a lecture by M. Koziar Vašáková)
Ethical ApprovalNo part of the contents of the manuscript has already been published in any other journal. All the authors agree to the reported contents and transfer their copyright to SEPAR.
Author ContributionsJG: writing the manuscript. MKV: concept and design of the study, data acquisition, writing the manuscript. YA and MS: data acquisition, writing the manuscript. MŠ, NM, MRK, MD, M P, VM, MS, MŽ, LL, KL, VB.: data acquisition. PO and OM: statistical analysis and interpretation. All authors contributed to the interpretation of the results and critical revision of the manuscript and approved the final draft.
FundingThe EMPIRE registry was supported by Boehringer Ingelheim and F. Hoffman-La Roche. The funders had no role in the design, analysis, or interpretation of the results in this study. The funders were given the opportunity to review the manuscript for medical and scientific accuracy, as well as intellectual property considerations.
Conflicts of InterestYA, MD, JG, MRK, LL, OM, PO, MP, MS, MŠ. Declare no conflict of interest. V. B. declares consulting fees, honoraria for lectures and advisory boards, travel support from Boehringer Ingelheim. K. L. declares grants, consulting fees, honoraria for lectures, travel support from Boehringer Ingelheim and Roche. N. M. declares consulting fees, honoraria for lectures, travel support from Boehringer Ingelheim and Roche. V. M. declares consulting fees from Boehringer Ingelheim and Roche. M. K. V. declares support of the manuscript from Boehringer Ingelheim, consulting fees, honoraria for lectures from Boehringer Ingelheim and Roche, honoraria advisory boards and travel support from Boehringer Ingelheim.
The authors thank investigators in clinical centres involved in the EMPIRE project for their valuable collaboration and data acquisition.
The following are the supplementary data to this article:







